先睹为快
靶点
人组织蛋白酶L(human cathepsin L),克鲁兹锥虫(Trypanosoma cruzi Cruzain)半胱氨酸蛋白酶
计算方法
分子对接
计算软件
MOE
单位与作者信息
美国Scripp海洋学研究所William H. Gerwick
发表杂志
Journal of Medicinal Chemistry
计算流程
将人组织蛋白酶L晶体结构(PDB code: 2XU3)中共价配体除去,使用MOEinduced fit对接模式,将天然产物Gallinamide A对接至活性位点Cys25附近。过滤掉烯酰胺的反应性碳与cys硫醇距离过远的结合模式,得到打分最高的结合模式(S=-10.70)并在此基础上进行结构修饰,发现活性更好先导化合物。
背景

长期以来,天然产物在药物发现中发挥着重要的作用,约70%的临床有效药物是受天然产物或其衍生物启发而得到的。近年来,海洋天然产物也成为了新药的来源之一,目前从海洋生物中提取出来用于临床的药物已达到13种且有31种处于临床开发阶段。半胱氨酸蛋白酶是治疗神经系统疾病和寄生虫病极好的潜在靶标,首次从巴拿马海洋蓝细菌中分离出来的化合物Gallinamide A被证实具有抗恶性疟原虫的活性,此后又陆续发现该化合物能与组织蛋白酶L不可逆结合,是人组织蛋白酶L的一种既有效且具有高度选择性的抑制剂。人组织蛋白酶L参与产生具有突触功能的肽神经递质的过程,在神经退行性疾病中起重要作用。经序列比对,它与从导致热带病的寄生虫中发现的许多蛋白酶具有高度的序列一致性和结构相似性,比如克鲁兹锥虫半胱氨酸蛋白酶(Trypanosoma cruzi),这种蛋白酶是恰加斯病的病原体,针对这种病的治疗方法可能会产生严重不良反应和感染。因此迫切需要开发针对造成慢性感染的无鞭毛体阶段的新疗法,针对人组织蛋白酶L的抑制剂能为抗南美锥虫病药的发展提供有用骨架分子。
分子对接模拟及理性药物设计
将人组织蛋白酶L和腈抑制剂共价结合的共晶结构导入MOE中,除去腈配体并修复蛋白结构,将Protonate 3D模块的PH值设为5.5(质子化蛋白酶L能达到的最大活性),力场选择Amber14:EHT。将Gallinamide A导入MOE,按同样的标准进行质子化和能量最小化。对接方案选择induced fit(诱导契合)将分子对接到处理后的组织蛋白酶L上,允许受体配体具有一定程度的柔性。计算烯酰胺的反应性碳与半胱氨酸硫醇中硫原子的距离,据此再过滤掉不合适的结合模式,并采用GBVI/WSA dG的结合自由能打分方法获得小分子的最优结合构象,最优构象对接得分为-10.70,与之前报导的姿势比较发现酰胺核心官能团的羰基在活性位点裂口的氧阴离子孔中通过形成氢键得到了良好的支撑,这是稳定中间抑制物质的重要特征。以此结合姿势作为起始结合姿势,对化合物1的结构进行了结构修饰得到16个化合物,能量最小化后重新进行分子对接,评定预测的结合亲和力和反应性碳和硫原子之间的距离变化。基于与常见组织蛋白酶底物的比较以及MOE对结合亲和力的预测,改变天然产物1中R’和R’’对应的L-Ala为L-Phe均能适度改善对接分数。越负的对接分数预计具有越高的结合亲和力。
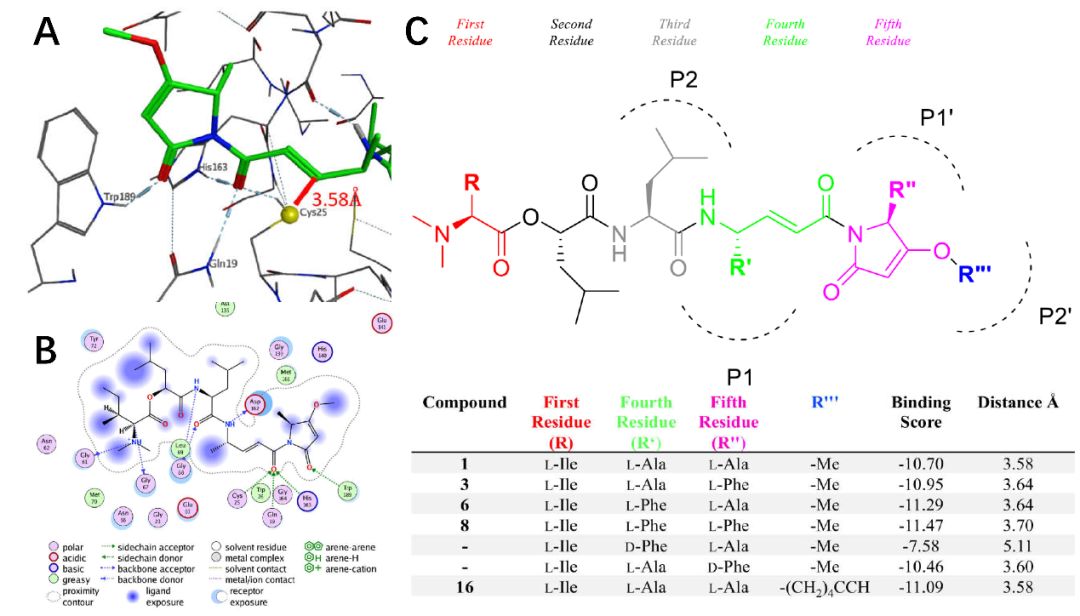
图1. 组织蛋白酶L和Gallinamide A的结合模式(A)和相互作用图(B), 对化合物1进行的结构修饰和对接打分(C)
图片来源JMC
Gallinamide A的全合成和及其酶抑制动力学分析
逆合成分析将分子分为3个部分:酰胺核,环化的头部和由脂肪族氨基酸组成的亲脂性尾巴。第一步由Boc-L-丙氨酸-OH形成酰胺核,首次反应形成Weinreb酰胺。头部的合成路线很大程度上取自Stolze的路线,该路线使用基于EDC的偶联反应附加丙氨酸并形成烯酰胺核心,随后将其脱保护,用Meldrum酸扩展并环化。通过Mitsunobu反应捕获所得的烯醇以完成头部基团。在形成尾部并将其耦合到酰胺核的最后步骤中,该路线也很大程度上遵循了Stolze的步骤,但进行了一定程度的修改。
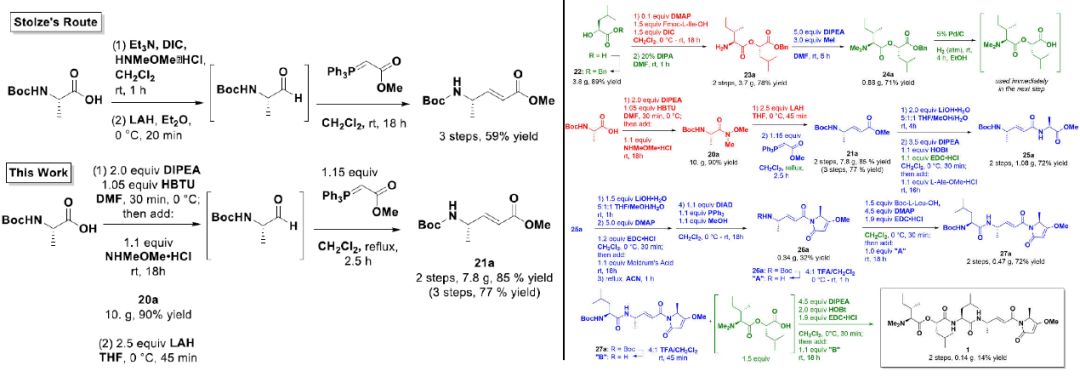
图2. Gallinamide A的全合成路线
图片来源JMC
不可逆抑制剂的IC50在抑制剂和酶浓度增加的情况下会呈现出随时间变化的趋势,这使得终点剂量反应实验不适用于评估抑制剂的效力。因此,作者采用动力学方法确定抑制的速率和效力。将抑制剂和酶与10μM的荧光底物cbz-FR-AMC混合,并在90分钟内监测产物的形成,产生的进度曲线的抑制剂浓度在0.0169-1000nM之间,将曲线基于两步不可逆反应拟合至非线性最小二乘回归曲线。计算出的Gallinamide A的Ki为4.67±0.40 nM,与先前报导的5.0nM一致。kinact/Ki = 901 000表明化合物1是一种非常有效的抑制剂。大多数化合物的kinact 非常相似,表明这些Gallinamide A衍生物中抑制剂的驱动力是可逆EI配合物的形成速率而不是共价键形成速率造成的。且将烯酰胺中的双键还原之后得到的化合物17,稀释实验显示17在1h后的动力学读数中表现出使酶活性恢复的迹象,证实了烯酰胺核心是天然产物的药效团。
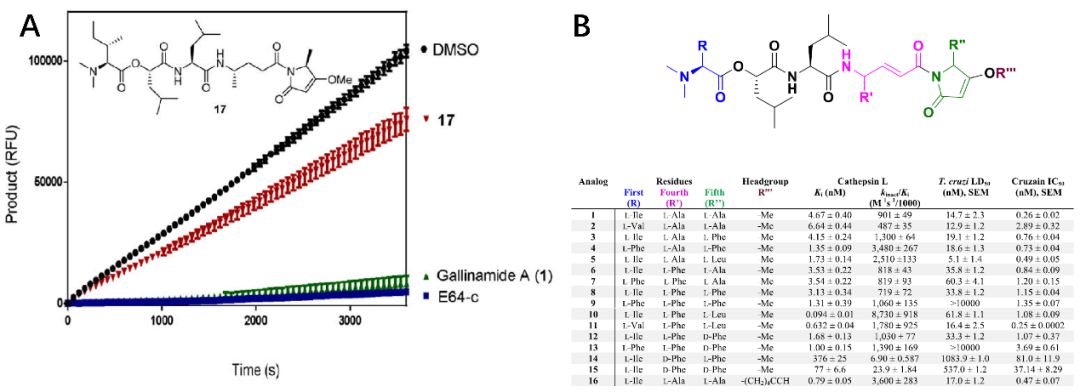
图3.化合物17的稀释实验及Gallinamide A(A)衍生物的动力学数据(B)
图片来源JMC
抗南美锥虫病的活性研究
基于组织蛋白酶L和克鲁兹锥虫蛋白酶之间结构和功能的相似性,用无鞭毛阶段的锥虫去测试Gallinamide A及其衍生物的生物活性。将锥虫感染的宿主细胞孵育3天之后添加待测试化合物,分析发现Gallinamide A显示出显著活性,LD50= 14.7 nM±2.3,并证实其对重组Cruzain的IC50达到0.26nM±0.02。化合物5是最有效的衍生物,LD50 仅为5.1±1.4 nM,对浓度高达10μM的宿主细胞无毒性。Cruzain对寄生虫的生存至关重要,因此,化合物对其的抑制将会迅速导致克鲁兹锥式原虫的细胞死亡。
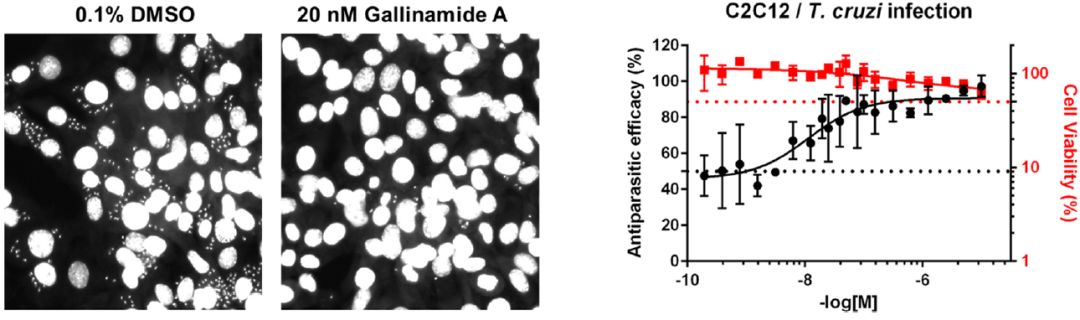
图4. Gallinamide A(1)可以有效地从小鼠宿主细胞中消除克氏锥虫
图片来源JMC
总结
本文利用分子对接和建模以及全合成的方法,确定了天然产物Gallinamide A中的几个结构特征,并对这些结构特征进行修饰得到16个衍生物来改善与人组织蛋白酶L的不可逆结合。动力学分析和烯烃饱和实验进一步确定了Gallinamide A发挥抑制作用主要是烯酰胺核心基团参与的不可逆步骤造成的。基于人组织蛋白酶L和克氏锥虫蛋白酶的结构功能相似性,抗南美锥虫病的活性研究证明Gallinamide A及衍生物5具有nM级别抗克氏锥原虫的活性,为抗南美锥虫病和可能的抗白热病药物的发展提供了有用的分子骨架。
参考文献
Boudreau PD, Miller BW, McCall LI, et al. Design of Gallinamide A Analogs as Potent Inhibitors of the Cysteine Proteases Human Cathepsin L and Trypanosoma cruzi Cruzain. J Med Chem 2019.