Introduction
Immunogenicity is an important factor affecting the success rate of clinical development of biological drugs. It is difficult to predict in vivo immunogenicity based on in vitro experiments. The existing prediction algorithms are generally ineffective with high false positives. AlphaMHC is a next gneration immunogenicity prediction algorithm proposed by Wecomput Technology, a compuation-driven company commited to drug discovery innovation.
Compared with prior arts, AlphaMHC has a newly designed implementation, providing a better solution for immunogenicity prediction. It is developed on top of more comprehensive understanding of the biological mechanism of immunogenicity, explores and integrates informative data from more dimensions and sources, and adopts state-of-the-art deep learning technologies. It effectively reduces false positives, has been verified against clinical data and has proved useful in tens of practical biologics R&D projects, which is highly recognized by our partners and customers.
Feature highlights
1.Significantly expanded training set space by collecting more data from literature and patents, besides all publicly available data sets, as well as from wet lab experiments of in-house or cooperation projects. The data types are mainly polypeptide affinity data, T cell activation data, proteomics data, antibody sequencing data, etc., with over 1 billion data entries/points.
2.Unlike most other algorithms which predict only the MHC-peptide binding affinity, AlphaMHC predicts the eventual immunogenicity at in vivo level, taking into consideration other important influencing factors besides peptide binding, such as immune tolerance, allele frequency, etc.
3.Single deep neural network is trained for up to 5000+ alleles of MHC-II. With the support of parallel computing, all supported MHC alleles can be simultaneously calculated in a high-throughput manner, while similar methods can usually only afford a few representative alleles within reasonable time cost.
4.An architecture achieving translational invariance in MHC-peptide binding analysis, making it suitable for identification of T-cell epitopes of va
5.riable lengths.
Performance assessment
To assess the prediction performance of AlphaMHC, we performed multiple validation tests against independent benchmark data sets without overlapping the training set.
Test 1 – MHC-peptide binding prediction
The performance of AlphaMHC in predicting peptide binding to MHC of class II is measured on a 5-fold cross-validation data (Refaeilzadeh et al., 2009), which is derived from IEDB (Vita et al., 2015). Each of the 5 test files consists of 13 000 data-points of MHC binding peptides. We compared AlphaMHC to a broadly known and widely used software NetMHCIIpan-3.2 (Jensen et al., 2018). As shown in Fig. 1, AlphaMHC outperforms NetMHCIIpan on both accuracy and AUC-ROC metrics.
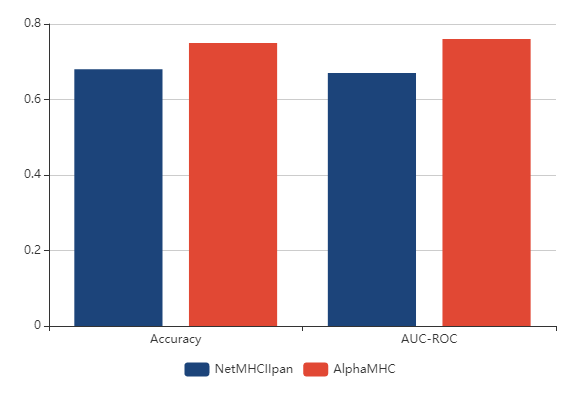
Figure 1. The accuracy and AUC-ROC of AlphaMHC and NetMHCIIpan on the peptide binding test set of MHC class II.
Test 2 – Immunogenicity prediction
To validate whether AlphaMHC could predict clinical outcomes of immunogenicty, we collected ~100 therapeutic antibodies that have been marketed or are in late clinical stage. According to the test, AlphaMHC can accurately categorize marketed therapeutic antibodies based on their predicted risk of immunogenicity. Chimeric, humanized, and fully human antibodies are predicted to have decreasing number of T cell epitopes, which is consistent with common sense that they have decreasing immunogenicity potential.
The result is shown in Fig. 2.
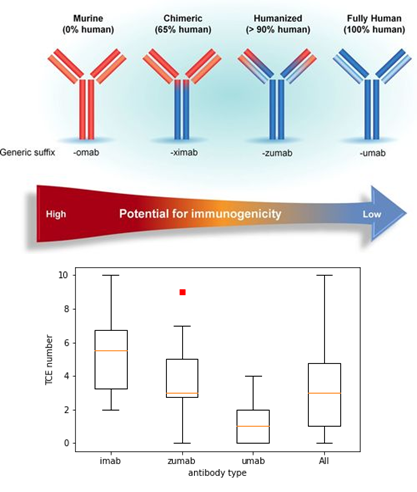
Figure 2. Predicted number of T-cell epitopes (TCE) of chimeric (imab), humanized (zumab), fully human (umab) antibodies. The higher the degree of antibody humanization, the lower the predicted risk of immunogenicity.
Moreover, we have collected several representative therapeutic biologics to form a typical test set for immunogenicity prediction evaluation, including mono/multi-specific antibodies and recombinant proteins. According to this test, AlphaMHC can accurately predict the immunogenicity risk of most entities, which correlates well with clinical outcomes, and show significantly better performance than the widely used NetMHCIIpan method (Table 1).
Table 1. Comparison of clinical immunogenicity and predicted risk for a collection of representative antibodies.

Practice:
Case demonstration
Case 1 – Natalizumab
Natalizumab (NZM), a humanized monoclonal IgG4 antibody to α4 integrins, is used to treat patients with relapsing-remitting multiple sclerosis (MS), but in about 6% of the cases persistent neutralizing anti-drug antibodies (ADAs) are induced leading to therapy discontinuation. To understand the basis of the ADA response and the mechanism of ADA-mediated neutralization, an in-depth analysis of the B and T cell responses was performed in two patients. In both patients, the analysis of the CD4+ T cell response, combined with mass spectrometry-based peptidomics, revealed a single immunodominant T cell epitope spanning the FR2-CDR2 region of the NZM light chain. Moreover, a CDR2-modified version of NZM (five de-immunized variants) was not recognized by T cells (Nature Medicine, 2019, 25, 1402–1407).
Using AlphaMHC, we scanned the full-length sequences of both heavy and light chain of NZM and its five de-immunized variants, and identified only one potential T-cell epitope (consisting of three overlapping peptide cores), right in the FR2-CDR2 region of NZM, which is shown in Table 2. This prediction is highly consistent with the experimental result.
Table 2. Comparison of AlphaMHC predictions with experimental results of Natalizumab and its de-immunized variants.
| Name | Experimentally validated T cell epitope |
AlphaMHC predicted T cell epitope |
| NZM | RLLIHYTSALQPGI (spanning the FR2-CDR2 region of the NZM light chain) |
LIHYTSALQPGI (Consisting of three overlapping cores LIHYTSALQ, IHYTSALQP, YTSALQPGI) |
| NZM var1 | None | None |
| NZM var2 | None | None |
| NZM var3 | None | None |
| NZM var4 | None | None |
| NZM var5 | None | None |
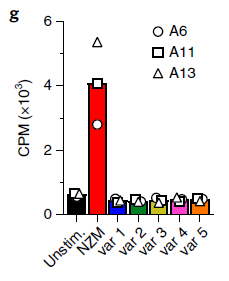
Figure S1. Proliferation of three NZM-LCFR2-CDR2-reactive T cell clones (A6, A11 and A13) after stimulation with autologous B cells pulsed with NZM and the five engineered variants (representative of n = 2 independent experiments). The bars show the mean proliferation. Source: Nature Medicine, 2019, 25, 1402–1407.

Figure S2. Sequence alignment of NZM and variants
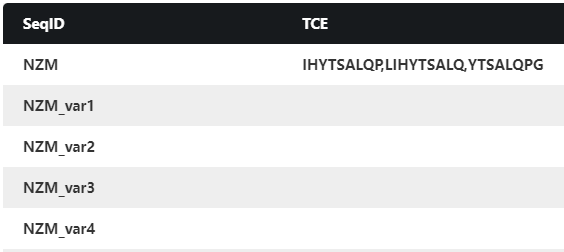
Figure S3. A snapshot of the output of AlphaMHC for NZM and de-immunized variants.
Case 2 – Factor VIIa
Vatreptacog alfa (VA), a recombinant human factor VIIa (rFVIIa) analog developed by Novo Nordisk to improve the treatment of bleeds in hemophilia patients with inhibitors, differs from native FVIIa by three amino acid substitutions (V158D, E296V or M298Q). In a randomized, double-blind, crossover, confirmatory phase III trial, 8/72 (11.1%) hemophilia A or B patients with inhibitors treated for acute bleeds developed anti-drug antibodies (ADAs) to vatreptacog alfa, which is terminated due to high immunogenicity. In two patients, for whom PK profiling was performed both before and after the development of ADAs, vatreptacog alfa showed a prolonged elimination phase following ADA development. During the follow-up evaluation, the rFVIIa cross-reactivity disappeared after the last vatreptacog alfa exposure, despite continued exposure to rFVIIa as part of standard care. Results from the vatreptacog alfa phase III trial demonstrate that the specific amino acid substitutions made to the FVIIa molecule alter its clinical immunogenicity. Unlike the bioengineered rFVIIa variants, there have been no reports of ADAs with the WT-rFVIIa, which has been used clinically for >2 decades. (J Thromb Haemost. 2015;13(11):1989-98).
A post hoc study demonstrated that the peptides with mutations at positions 296 and 298 are neo-epitopes, because they bind to human HLA-DRB1 alleles with high affinity, were identified on HLA-DRB1 molecules after processing by antigen presenting cells and resulted in T-cell activation, as shown in Fig. 3 (Sci Transl Med. 2017, 9(372); Blood Adv 2019, 3 (17): 2668–2678).
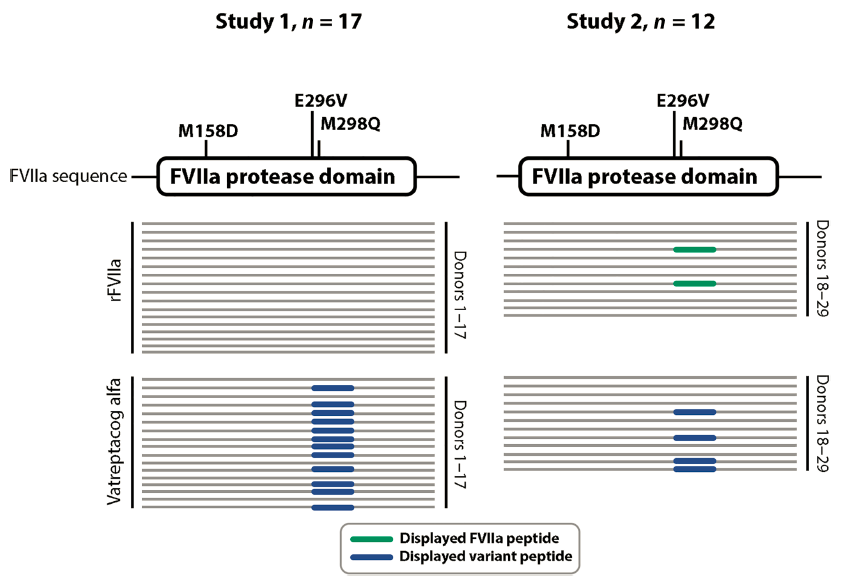
Figure 3. In vitro profiling of vatreptacog alfa–derived neoepitopes displayed by HLA-DR after uptake by DCs.
Schematic representation of data generated in two independent studies performed using (A) a 17-donor cohort and (B) a 12-donor cohort. Each line represents one hit or peptide identified by MS. The mutations introduced in the protease domain of FVIIa to generate vatreptacog alfa are indicated in the figure. The FVIIa peptides identified are illustrated in gray, andmutant vatreptacog alfa peptides are illustrated in black. Source: Sci Transl Med. 2017, 9(372).
Using AlphaMHC, we scanned the full-length sequences of VA, and identified only one potential T-cell epitope (consisting of six overlapping peptide cores), right in the E296V/M298Q region, which is shown in Table 3. Also, wild type FVIIa is predicted to have no T-cell epitopes. This prediction is highly consistent with the experimental result.
Table 3. AlphaMHC predictions and the experimental results of WT-FVIIa and VA.
| Name | Experimentally validated T cell epitope |
AlphaMHC predicted T cell epitope |
| WT-FVIIa | None | None |
| VA(rFVIIa analog) | DRGATALVLQVLNVPRL
(spanning the E296V/M298Q region) |
LDRGATALVLQVLNVPR
(Consisting of six overlapping cores:ATALVLQVL, DRGATALVL, GATALVLQV, LDRGATALV, LQVLNVPRL, VLQVLNVPR) |
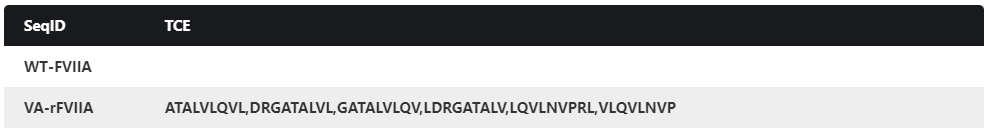
Figure S4. A snapshot of the output of AlphaMHC for wild-type FVIIa and vatreptacog alfa.
Case 3 – Ixekizumab
Ixekizumab is a humanized IgG4 variant/kappa antibody, obtained by immunizing mice with human IL-17A, selecting specific binders using antigen-binding fragment (Fab)-expressing phage display technology, followed by phage display-based humanization and affinity maturation in E. coli. Ixekizumab is the second IL-17A monoclonal antibody (mAb) approved for the treatment of moderate to severe plaque PsO and PsA. In a pooled analysis of three Phase 3 studies involving patients with psoriasis, ADA against ixekizumab developed in 9% of patients by Week 12, and those patients with higher titers of ADA experienced a reduction in clinical efficacy. According to the product label, approximately 22% of patients treated with ixekizumab develop antibodies during the 60-week treatment period. Ixekizumab were further examined regarding the specific T cell epitopes. ixekizumab-specific CD4 T cell lines were generated from 31 healthy, treatment-naïve donors via 28-day co-culture with mature monocyte-derived dendritic cells exposed to the antibody. 32 T cell lines from eight donors were obtained for ixekizumab. For 11 of these T cell lines, the specific T cell epitopes could be identified and confirmed by major histocompatibility complex–associated peptide proteomics as being naturally presented peptides. most of which are in the light chain. Most of the T cell epitopes are identified in ixekizumab light chain, as shown in Fig. 4 (mAbs, 2020, 12:1, 1707418).
Using AlphaMHC, we scanned the sequences of ixekizumab, and identified 3 potential T cell epitopes consisting of five overlapping peptide cores, corresponding to CDR1, CDR2, and CDR3, covering most non-germline CDR residues (Fig. 4). In general, the prediction is consistent with the experimental results.
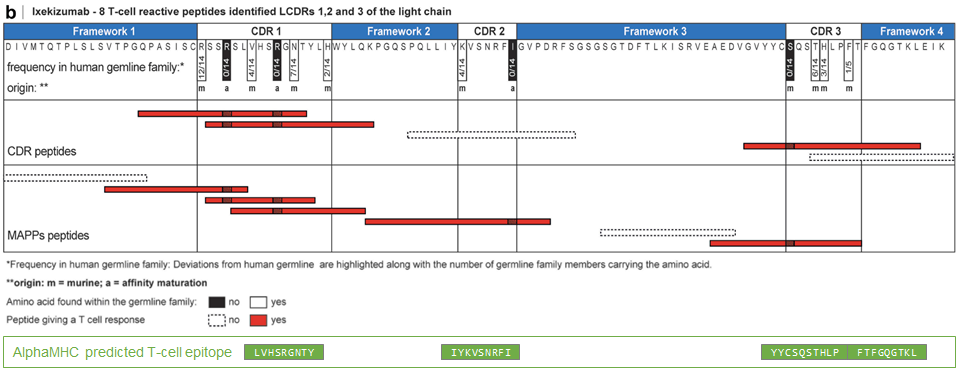
Figure 4. T cell epitopes identified in ixekizumab light chain.
CDR and MAPPs derived peptides are mapped graphically below the amino acid sequence and colored according to their reactivity in the T cell assay. Ixekizumab light chain were aligned to closest germline IGKV2D-29*02|IGKJ2*01. Deviations in the amino acid sequence are labeled according to their origin and highlighted along with the number of germline family members. MAPPs, major histocompatibility complex–associated peptide proteomics. Source: mAbs, 2020, 12:1, 1707418. Additionally, AlphaMHC predicted T-cell epitopes are mapped graphically in the bottom. The predicted epitopes have good correlation with experimentally identified ones.