先睹为快
靶点
A3腺苷受体adenosine A3receptor (A3R)
小分子配体
激动剂分子NECA和IB-MECA
计算方法
同源模建(homology modeling)
分子对接(Docking)
分子模拟(MD Simulation)
自由能计算(MM-GBSA Calculations)
计算软件
Adenosiland web-service,GOLD/ChemScore,Desmond,Schrodinger
单位与作者信息
剑桥大学的Graham Ladds教授;
雅典大学的Antonios Kolocouris教授
发表杂志
Journal of Medicinal Chemistry
计算流程
模建A3R的蛋白结构,将激动剂对接到活性口袋中,选择每个模型中打分top3的binding poses进行150ns的MD模拟,轨迹分析A3腺苷受体与激动剂的相互作用特征。关键的氨基酸进行突变实验验证,并与MM-GBSA计算结果ΔGeff进行对比,揭示影响激动剂活性的重要残基,用于后续A3R激动剂的设计指导
背景
腺苷受体(ARs)是G蛋白偶联受体(GPCRs)的家族成员,迄今为止,已经鉴定出A1, A2A, A2B, A3四个亚型。本文研究的A3腺苷受体(A3R)被证实在各种肿瘤细胞中的高表达,可以作为抑制癌细胞增殖的药物靶点。腺苷(Ado)是A3R的内源性激动剂,通过对Ado的核糖部分修饰和腺嘌呤环取代,发现了两类激动剂NECA和IB-MECA(图1)。其中,NECA分子是一类非选择性AR激动剂,由于对各种亚型的腺苷受体都有作用;IB-MECA是目前最有效的亚型选择A3R激动剂,已经处于炎症和癌症的临床试验。
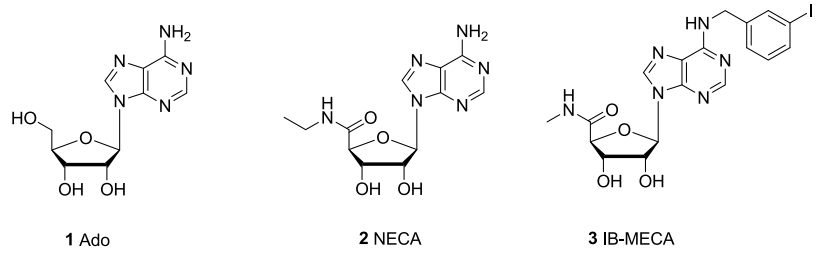
图1. 腺苷分子,激动剂NECA和IB-MECA的结构示意图
图片来源JMC
腺苷受体亚型中A1, A2A 和Ado/激动剂的复合物晶体已经解析出来了,而A3R由于结晶困难,目前还没有晶体结构报道。所以,本文采用同源模建的方式,构建了激动剂和A3R的复合物模型,并对A3R进行了一些定点突变研究,以检验跨膜(TM)螺旋结构域TM3、TM5、TM6、TM7和第二细胞外环2(EL2)对激动剂识别和结合的重要性。通过分子动力学模拟和MM-GBSA结合自由能计算,揭示了激动剂与A3R复合物中尚未解决的重要结构特征。

计算方法详解
1
模建野生型和突变型的A3R-NECA复合物结构
野生型A3R结构通过Adenosiland服务器,以NECA-A2AR晶体结构 (PDB ID 2YDV)作为模板进行同源模建。首先,删除A2A蛋白的溶菌酶/抗体结构域,通过YASARA软件重构EL2和LL3结构域,在amber99sb 力场下对蛋白结构能量优化,直到达到共轭梯度小于0.05 kcal·mol−1·Å−1。优化后的A2A蛋白作为模建A3R的模板。在模建非保守区域前,先将模板的骨架原子和保守残基的坐标保留在新的结构中,使用amber99sb力场对得到的结构最小化,直到达到共轭梯度小于0.05 kcal·mol−1·Å−1 ;在分子操作环境(MOE)中对缺失的loop结构进行重构,同时将N-和C-末端截断,使其长度不超过晶体结构的长度,残基被重新质子化态。最后,根据OPM数据库将模型插入到POPC双层膜中,利用CHARMM27力场对模型进行了45ns的MD模拟。
接下来,通过薛定谔软件的蛋白准备页面,对A2A R-NECA和A3 R-NECA复合物结构优化,在这个过程中,分配键序和二硫键,并添加了缺失的氢原子。此外,蛋白质模型的N-末端和C-末端分别连上乙酰基和N-甲基-氨基。使用OPLS2005力场对蛋白质进行全原子最小化,重原子均方根偏差(rmsd)值限制在0.30Å。旋转A3 R-NECA 复合物的V169残基的侧链,来增加嘌呤环6-NH2取代的激动剂自由空间。通过野生型的A3 R-NECA 复合物结构,构建18个突变型的A3 R-NECA复合物结构。
2
选择最优的对接方法
为了选择一种适合A3R的分子对接方法,分别测试了GOLD的ChemScore和GoldScore打分函数,Glide的Glide SP 和 Glide XP打分函数,能否准确重现NECA在A2A R蛋白晶体中的结合构象。结果显示,Glide的大部分docking pose中NECA的核糖结构会错误地朝向结合口袋的溶剂暴露区;相反,GOLD产生的大部分docking pose中NECA的核糖结构朝向蛋白内部,并且使用ChemScore打分函数,这种docking pose所占比例最高(超过80%)。最终,选择了GOLD/ChemScore方法对接NECA,IB-MECA到19个A3 R蛋白,每个复合体的三个得分最高的docking poses用于MD模拟。
3
MD模拟
为了选择合适的力场,分别在OPLS005力场、amber99sb力场和CHARMM27力场下,通过300 ns的分子动力学模拟,测试了POPE双层膜中NECA-A2AR的复合物的稳定性。结果显示amber99sb力场表现得更好。
使用Desmond V4.9的System Builder程序将蛋白质-配体复合物插入预平衡水合膜双层(POPE)中,POPE 双分子层包含160种脂质,15000个Tip3p水分子,在水相中加入钠和氯离子,中和体系电荷,正交周期盒边界为15Å,原子总数约为70 000。
用desmond软件和amber99sb力场对19个A3R-NECA和19个A3R-IB-MECA复合物进行了150ns的MD模拟。首先是对蛋白质周围的溶剂和离子进行平衡,在10K温度,NVT系综,进行200ps的限制性动力学模拟,对蛋白质中的重原子(非氢原子)施加50 kcal·mol−1限制力,保障平衡蛋白质周围的溶剂分子时, 而不引起蛋白质结构的变化。然后,在NPT系综下,将每个体系在200ps内从 10K 逐渐升温到 310K,此时溶剂水分子采用同样的力进行限制。升温完成之后,对体系进行2ns的NPT平衡模拟,慢慢放开限制, 让体系弛豫到新的状态。完成两个阶段的预平衡后, 体系在需要的压力与温度下平衡好了,现在可以放开位置限制,进行150 ns的成品MD模拟。
在分子动力学模拟过程中,Particle-Mesh-Ewald(PME)方法用来处理长程静电作用,SHAKE 算法则被用于限制所有含氢的键,同时,设置范德华作用和短程静电作用的截断值为 9Å。利用maestro的图形用户界面对产生的轨迹进行可视化,并利用desmond提供的模拟相互作用图(sid)工具进行蛋白质-配体相互作用分析。
4
MM-GBSA 计算
对每个体系最后50ns的轨迹进行能量分析,采用薛定谔软件的相关模块中MM-GBSA方法评估小分子的结合力。在计算之前,所有的水分子、离子和脂类都被去除,并将蛋白复合物重新放置回几何中心。每个复合物的有效结合自由能计算通过thermal_mmgbsa.py脚本实现,本文中的结合自由能值对应于复合体的三个最高得分binding poses的MD模拟中获得的平均值±标准偏差。
结果部分
MD模拟结果表明,激动剂通过氢键、范德华和π-π相互作用结合到A3腺苷受体的活性口袋中。如图2所示, T94、S271、H272、N250残基都可以与IB-MECA形成氢键相互作用;F168与IB-MECA的嘌呤芳环形成π-π相互作用;L246、I268、W185、W243、V169、L264与IB-MECA形成范德华相互作用。上述残基除了V169、L264外,在NECA激动剂与A3腺苷受体复合物中也有类似的作用特征。
结合突变实验,发现激动剂结合中关键的残基,并用MM-GBSA方法计算38个A3RS复合物中激动剂的结合自由能ΔGeff,发现导致激动剂活性降低的突变体的能量值明显高于维持或增加激动剂活性的突变体,并且与实验测定的pIC50值具有显著的相关性(表1)。
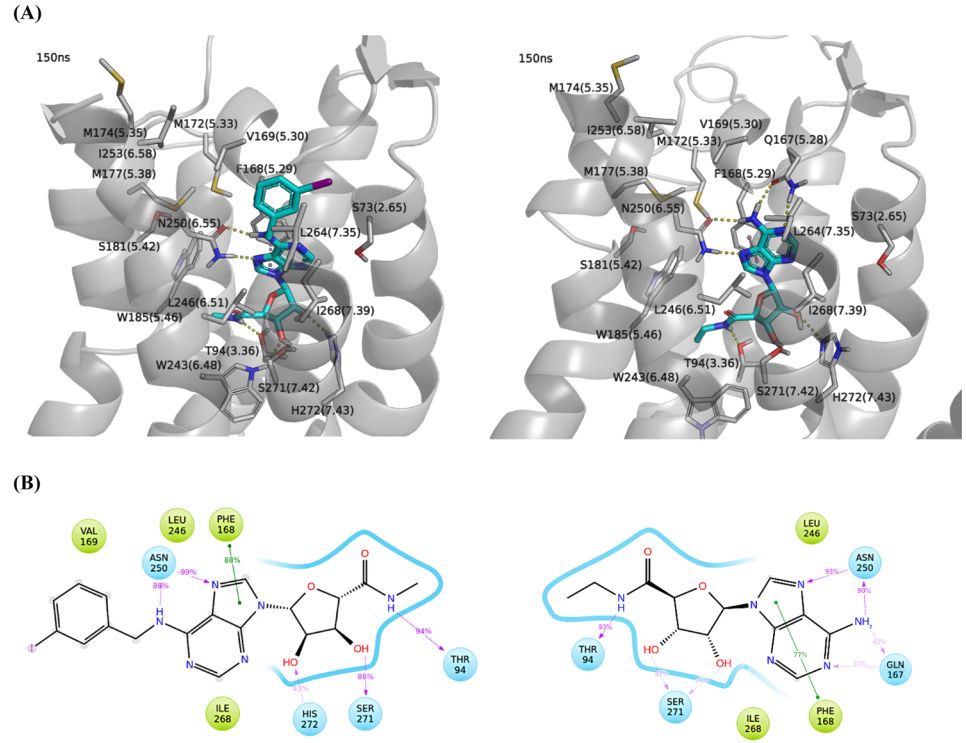
图2. IB-MECA(左)和NECA(右)与A3腺苷受体复合物结构图和二维相互作用图
图片来源JMC
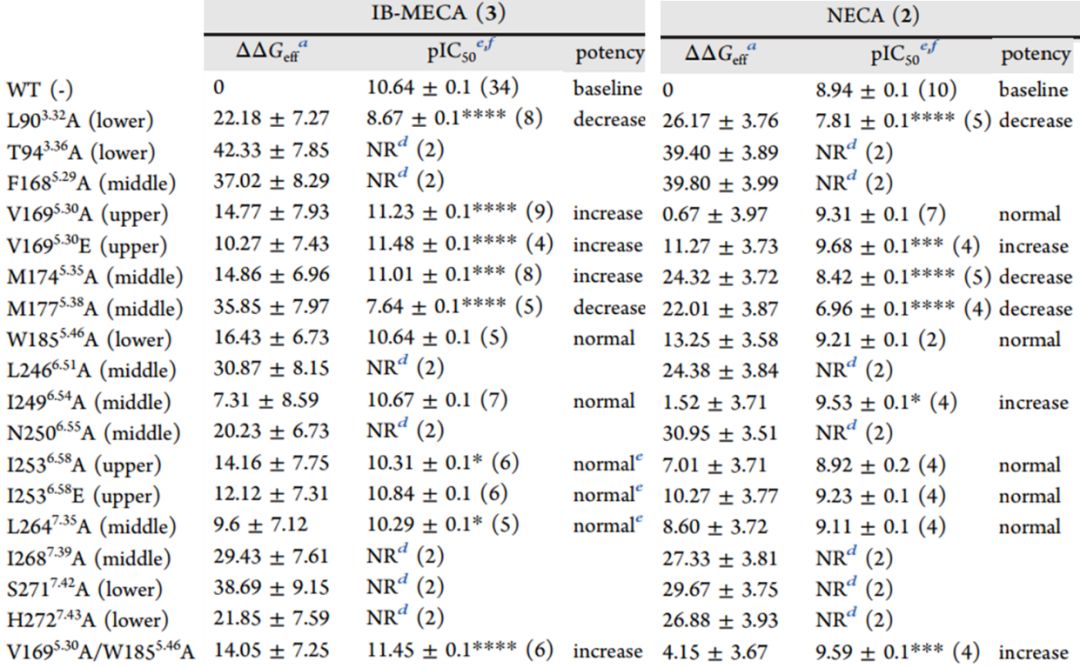
表1. IB-MECA、NECA激动剂与A3腺苷受体复合物的活性pIC50值以及结合能ΔGeff
图片来源JMC
与受体有直接作用的残基有T94、F168、L246、N250、I268、S271、H272,这些残基突变成丙氨酸会降低激动剂活性;W185A、I264A突变对活性没有影响;比较特别的是V169残基,当突变成丙氨酸会增加IB-MECA激动剂的活性,当突变成谷氨酸对两种激动剂都有增强作用;间接作用的残基如L90、 M177、 M175、 I249 、I253对激动剂有双重作用。
在许多情况下,简单的轨迹分析足以解释复合物的稳定性,然而一些降低激动剂活性的突变没有明显改变配体的结合构象,如H272A,通过计算ΔGeff,发现结合能高于野生型22 kcal mol−1,说明突变体的配体结合力变弱了,解释了活性降低的原因。表明MM-GBSA计算的ΔGeff可以作为区分活性和非活性复合物的参考。
总结
本文1研究发现,F168的π-π相互作用, L246和I268的范德华相互作用是配体结合和激活受体的必需条件。同样,A3腺苷受体的活化通过与T94、N250、S271形成氢键作用来介导的;残基H272会影响激动剂的活性,但不影响与受体的结合;V169并不是IB-MECA激动剂的A3R选择性的来源,相反,结合口袋远端的 EL2, TM5和TM6的残基L90、M177,可能通过调节口袋的结构,影响激动剂的活性,这对未来设计强效和选择靶向A3R的激动剂具有重要指导意义。
参考文献:
Stamatis, D.; Lagarias, P.; Barkan, K.; Vrontaki, E.; Ladds, G.; Kolocouris, A., Structural Characterization of Agonist Binding to an A(3) Adenosine Receptor through Biomolecular Simulations and Mutagenesis Experiments. Journal of Medicinal Chemistry 2019, 62 (19), 8831-8846.