

引言
癌症特异性代谢改变过度激活了哺乳动物雷帕霉素靶蛋白(mTOR)的激酶活性,以克服压力环境。雷帕霉素能够变构抑制mTOR复合物1(mTORC1),已经被批准用作抗癌治疗药物。然而,雷帕霉素类化合物的免疫抑制副作用会促进肿瘤转移,从而限制了它们的治疗效果。在此,本文作者通过计算机虚拟筛选结合细胞实验首次报道了一种nonrapalog抑制剂——WRX606。文章研究表明,WRX606与mTOR的FK506结合蛋白12(FKBP12)和FKBP-雷帕霉素结合结构域(FRB)形成三元复合物,导致mTORC1的变构抑制。WRX606不仅可以抑制核糖体蛋白S6激酶1(S6K1)的磷酸化,还可以抑制eIF4E结合蛋白1(4E-BP1)的磷酸化。因此,WRX606能够在不促进肿瘤转移的情况下有效地抑制小鼠的肿瘤生长。这些结果表明,WRX606是一种有效的先导化合物,可以进一步用于开发新的抗癌药物。
背景介绍
代谢重编程或癌症特异性代谢改变是由癌细胞中的致癌信号介导的。癌细胞中的异常信号会加速mTOR复合物1 (mTORC1)和mTOR复合物2(mTORC2)的活动,以克服压力性的癌症微环境。mTORC1通过eIF4E结合蛋白1(4E-BP1)和核糖体蛋白S6激酶1(S6K1)的磷酸化来控制蛋白质合成和细胞增殖,以响应营养物、能量和生长因子的环境水平。mTORC2通过磷酸化蛋白激酶AKT、PKC和SGK来调节细胞存活、细胞周期和细胞骨架组织。
雷帕霉素(rapamycin)是从吸水链霉菌中分离出来的一种抗真菌大环内酯类药物,后来发现它可以通过变构抑制mTORC1来抑制哺乳动物的肿瘤生长。然而,临床前试验表明,这种对mTORC1或mTORC2的变构抑制作用很快会被耐受。与雷帕霉素一样,催化抑制剂在长期给药过程中也存在典型的缺点,即可能导致mTORC2激活。此外,同时兼具PI3K和mTOR双重抑制模式的抑制剂,如PI-103,对正常细胞显示出细胞毒性,表明其治疗范围有限。因此,需要一种新的抑制剂来解决当前mTOR抑制剂的缺点。
本文研究旨在结合使用计算机和细胞筛选的混合策略发现mTORC1的新变构抑制剂。基于雷帕霉素与mTOR的FKBP12和FRB相结合的复合物的晶体结构,作者通过分子对接对来自ZINC15数据库的虚拟文库进行了虚拟筛选,并通过基于分裂荧光素酶的细胞测定进一步分析结果,最终共鉴定得到了13种具有共同骨架的类似物。13种化合物之一的WRX606可以很好地与mTOR的FKBP12和FRB进行三元络合。此外,作者使用拉伸分子动力学(SMD)和基于细胞的点突变实验进一步阐明了WRX606进行三元络合的分子机制,结果表明WRX606的喹唑啉-2,4-二酮和丁酰胺基团与FKBP12形成氢键,而苯并二恶醇和3-氯苯氨基通过疏水相互作用与FRB相互作用。正是这些相互作用促成了FKBP12-WRX606-FRB三元复合物的形成,从而变构抑制了mTORC1,进而达到了抑制肿瘤生长的目的。相关研究以“In Silico and In Cell Hybrid Selection of Nonrapalog Ligands to Allosterically Inhibit the Kinase Activity of mTORC1”为题发布在著名药物化学杂志JMC上。
FKBP12/FRB结合配体的初步筛选
基于物理特性,从ZINC15数据库中制备了三个不同的虚拟配体库子集(L#1、L#2 和 L#3,总共659417个化合物)。所有对接均使用FKBP12-雷帕霉素-FRB三元复合物晶体结构(PDB:1FAP)在ICM-Pro软件上进行(图 1A)。雷帕霉素分子被用作对接模板,使用原子属性场(APF)引导配体结合到对接位点。通过用丙氨酸替换一对侧链的方法(扫描和优化,SCARE方法)引入了对接袋中的侧链灵活性,以提高对接的精度。结果表明,L#3(Mw > 500 Da)的对接得分通常比L#1和L#2中分子量较小配体的分数要好(图1B)。此外,使用ICM化学性质计算器对排名前三的配体,即来自L#2的ZINC32928513(WRX513;图1C)和来自L#3的ZINC100492939(WRX939;图1D)和ZINC8593606(WRX606;图1E)进行泛分析干扰化合物过滤(PAINS),结果表明它们都是非PAINS干扰化合物,并显示出一定程度的药物相似性。进一步使用NanoBiT(图1F)进行细胞测定,结果表明WRX606诱导了三元复合物的形成,而WRX513和WRX939则没有(图1G)。需要强调的是,在对接模型中,这三种配体以不同的模式与FKBP12和FRB相互作用(图1H-J)。
WRX606类似物的选择
使用WRX606的通用骨架,作者进一步鉴定了12种类似物(图2A)。所有这些类似物也是非PAINS干扰化合物。使用NanoBiT分析发现另一种类似物WRX601也可以增强三元复合物的形成,但其亲和力比WRX606弱(图2B)。进一步的定量分析(即通过细胞内检测系统对WRX606、WRX601和雷帕霉素进行滴定)结果表明WRX601比WRX606结合FKB12/FRB的能力弱两倍(图 2C)。分析对接模型发现,由于构象变化,WRX601没有与Y2104结合,从而导致在FKBP12位点缺乏与D37相互作用的氢键(图2D-F),进而导致上述差异。
最后,为了验证WRX606和WRX601的对接模型,作者在它们与FRB和FKBP12 的预测结合残基处进行了点突变(图3A-F)。其中,D37A、I56A和Y82A的点突变位点位于FKBP12的配体结合口袋中;S2035A、Y2038A、F2039A、T2098A、W2101A、Y2104A和Y2105A位于FRB的配体结合口袋中。可以看到,D37A、Y2038Y和Y2104A的点突变对WRX601结合的影响小于WRX606,表明当3-苯氨基位置(WRX606)上的氯原子被一个氢原子取代后,配体的结合模式就会不同。总体而言,点突变实验验证了对接模型,表明WRX606在细胞水平上比WRX601更容易与FKBP12和FRB形成三元复合物。作者最后还分别验证了WRX606作为S6K1和4E-BP1的变构抑制剂在癌细胞中发挥的主要作用,结果表明WRX606能够抑制S6K1和4E-BP1两种底物的磷酸化,而雷帕霉素和WRX601仅微弱地抑制4E-BP1的磷酸化。此外,WRX606在任一激活条件下对癌细胞均显示出显著的细胞毒作用,比对非癌细胞强100-1000倍,表明WRX606具有较好的抑制癌细胞生长的效果。
图表汇总

图 1. 虚拟筛选发现与FKBP12和FRB形成三元复合物的配体。图片来源于JMC
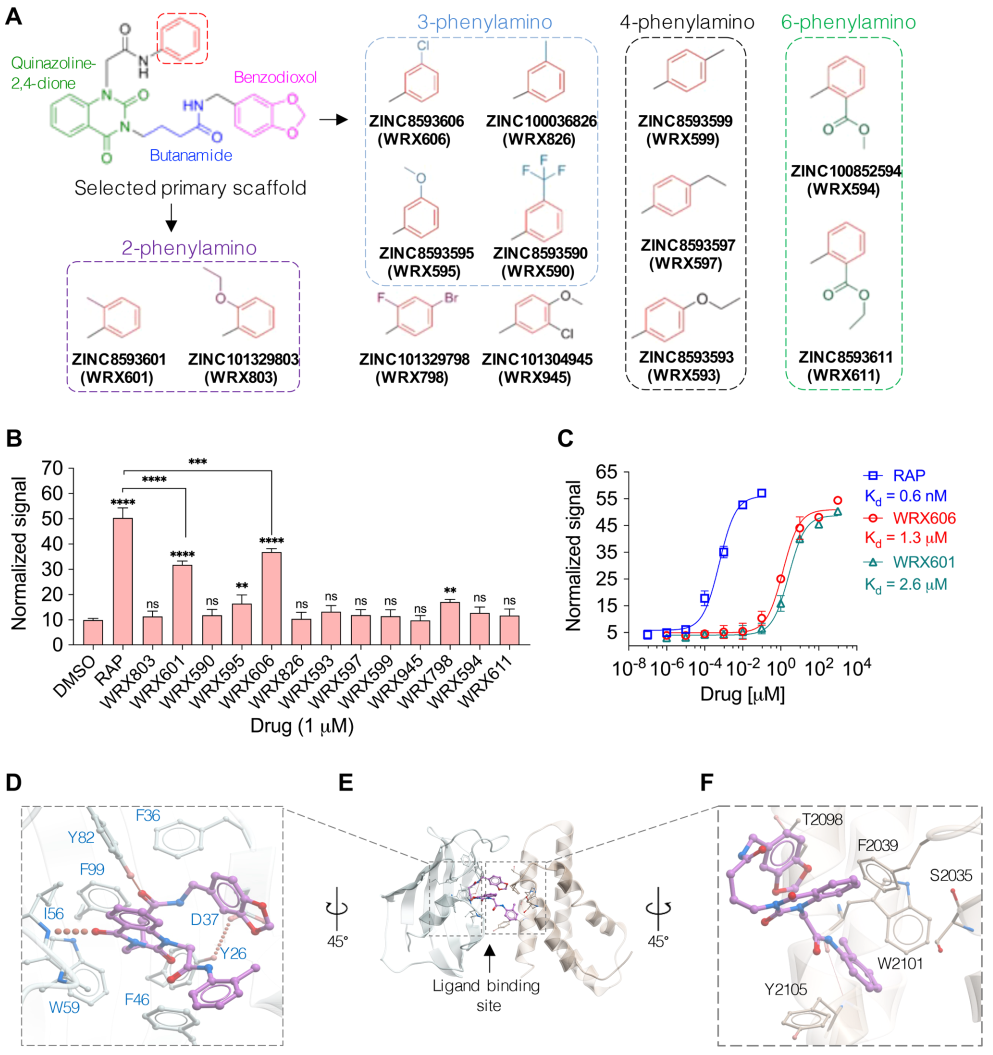
图2. 基于通用骨架选择WRX606类似物。图片来源于JMC
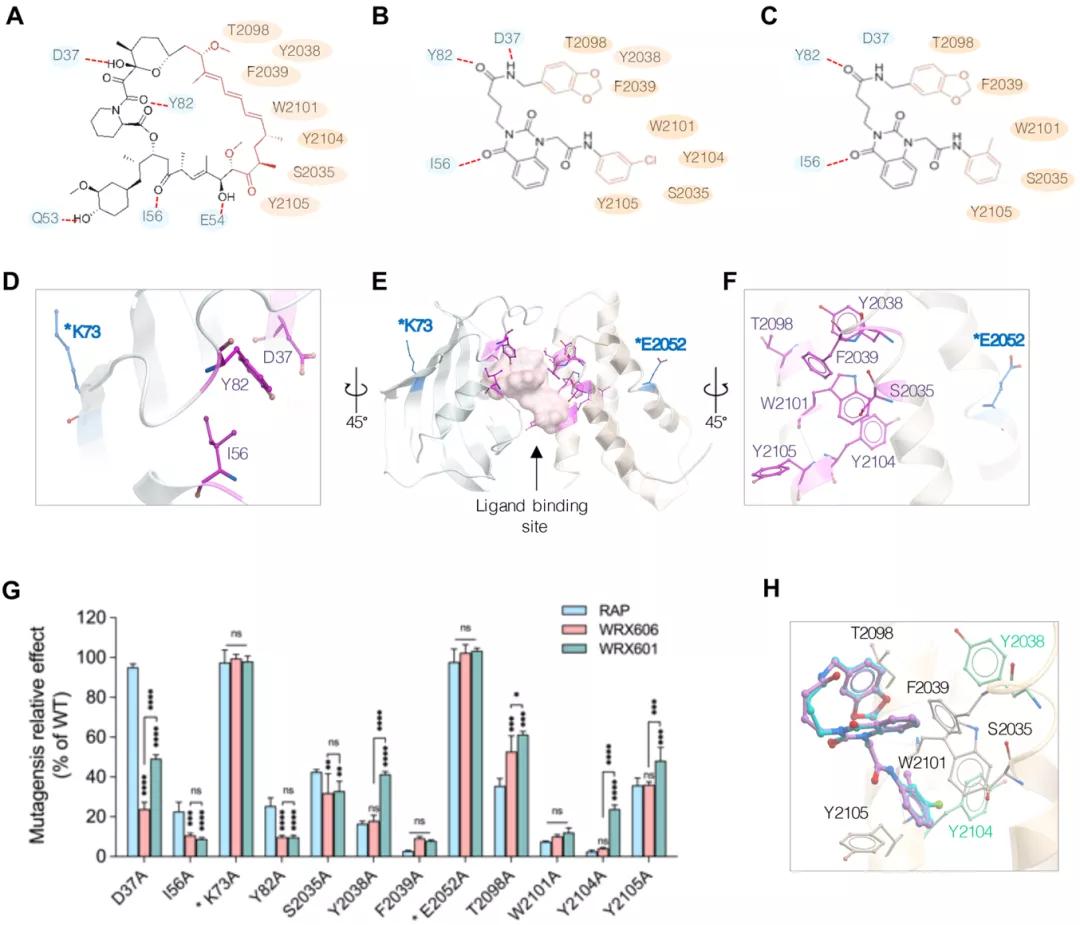
图3. WRX606与FKBP12/FRB界面的结合比WRX601更强。图片来源于JMC
结论总结
mTOR已被用作各种癌症的治疗靶点。本文通过计算机和细胞混合筛选的策略发现了一种非雷帕霉素类变构抑制剂——WRX606,其通过同时靶向mTORC1的FKBP12和FRB来实现配体选择。因此,所选化合物WRX606是第一个通过与FKBP12和FRB形成三元复合物而变构抑制mTORC1的非雷帕同系化合物。然后,作者证明WRX606能够显著抑制mTORC1的底物S6K1和4E-BP1的磷酸化,进而导致癌症细胞毒性。总之,这种新型的mTORC1变构抑制剂扩展了进一步发现和开发专门针对mTORC1的抗癌疗法的平台。
参考文献
Shams R, Matsukawa A, Ochi Y, Ito Y, Miyatake H, In Silico and In Cell Hybrid Selection of Nonrapalog Ligands to Allosterically Inhibit the Kinase Activity of mTORC1, J. Med. Chem., 2021, ASAP. DOI: 10.1021/acs.jmedchem.1c00536.