:JCTC | Chembot:一种实现选择性组态相互作用的机器学习方法")
:JCTC | Chembot:一种实现选择性组态相互作用的机器学习方法")
背景介绍
随着机器学习(ML)软件(scikit-learn、TensorFlow、PyTorch等)的广泛使用以及图形处理单元成本和速度的不断提高,ML作为一种预测工具变得非常流行并在诸多方面获得了成功。在过去的十年中,ML已经应用于量子物理和化学的各种原子模拟工作中。这些工作主要包括以下几个方面:1)用于材料设计或发现的化学空间探索;2)用于分子动力学的力场函数设计;3)密度泛函理论(DFT)的函数设计;4)相关能量预测。此外,也有使用ML模型本身来表示波函数的应用,如使用神经网络来描述电子的多组态相互作用。或者使用ML来构建波函数,如使用神经网络来进行选择性组态相互作用(SCI),各种SCI方法对组态选择有不同的标准,但其核心都是解决所选组态的变分空间中的特征值问题。SCI方法通常依赖于某种形式的扰动理论来执行选择。ML为选择组态的扰动理论提供了一种不同的替代方案,从而产生一种与现有SCI截然不同的SCI方法。基于此,在本文中作者构建了Chembot方法,一种实现选择性组态相互作用的机器学习方法。
主要内容
来自劳伦斯利弗莫尔国家实验室的Sergio D教授提出了一种基于机器学习的选择性组态相互作用方法,称为Chembot,它利用了许多新的选择方法进行了模型的设计和训练。这些选择包括:使用支持向量机(SVN)选择重要的组态,使用电荷密度矩阵和组态能量作为特征,使用启发式算法来提高训练数据的质量。该文章使用了斯莱特行列式并在MolPro Version下进行了计算。通过测试Chembot进行组态相互作用的能力,发现了该方法在更少的迭代收敛、更少的变分空间中的行列式以及更少的重要组态下却可以计算相同的能量,绝对优于基于蒙特卡罗的组态相互作用。此外该文章阐述了Chembot的原理及方法,并进行了简单的测试及应用。相关的成果以 “Chembot: A Machine Learning Approach to Selective Configuration Interaction” 为题发布在著名理论计算期刊J. Chem. Theory Comput.上。
图表汇总
:JCTC | Chembot:一种实现选择性组态相互作用的机器学习方法")
图1. Chembot算法的示意图。图片来源于JCTC
:JCTC | Chembot:一种实现选择性组态相互作用的机器学习方法")
图2. 特征向量分析图。图片来源于JCTC
:JCTC | Chembot:一种实现选择性组态相互作用的机器学习方法")
表1. H4的基准方法和FCI之间的能量误差。表格来源于JCTC
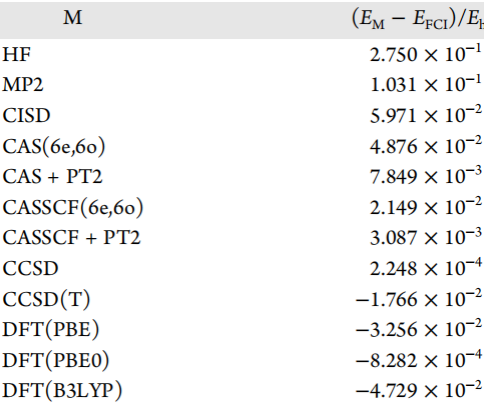
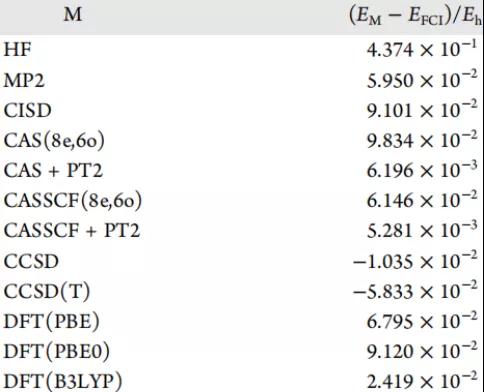
表3. 水的基准方法与FCI溶液之间的能量差。表格来源于JCTC
推荐理由
该文章开发了一种名为Chembot的基于ML的SCI方法,可以有效地选择重要的组态迭代地构建波函数。该方法使用SVM作为ML模型,使用电荷密度矩阵和能量作为训练特征、并通过启发式方法对数据进行训练。该方法的亮点在于它使用CAS波函数作为初始训练集而非CISD波函数,其波函数的计算也是基于Epstein-Nesbet微扰理论。此外,该文章的测试结果表明Chembot在组态选择效率等方面均具有良好的表现。
参考文献
Sergio D. Pineda Flores, Chembot: A Machine Learning Approach to Selective Configuration Interaction, Journal of Chemical Theory and Computation, 2021, ASAP. DOI: 10.1021/acs.jctc.1c00196.