众所周知,药物是可以在体内发挥生物活性作用达到治疗疾病目的的化合物,大部分药物的生物活性都是通过药物分子与体内的蛋白等生物大分子结合来介导的。[1]化合物的靶点结合作用机制是药物研发的重要理论基础,然而目前有大量的生物活性分子其作用靶点是未知的。此外,随着基因组学发展,越来越多的新型靶点正在被发现,老药新用也依赖于对这些药物新靶点的预测。[2]因此,化合物靶点预测具有重大科学意义。
目前靶点预测的方法有很多,大部分的文献都把这些方法分为实验和计算两大类。[3]实验方法中最直接的就是生化测试法,通过孵育小分子化合物和相应蛋白的混合物,洗脱后直接评估两者结合能力来判断小分子是否结合于靶点。[4]另外还可以利用基因组的方法,比如将小分子的作用靶点锁定在一定范围内的基因簇后,通过对该基因簇进行一系列点突变或敲除,如果突变或敲除的样本表现出和小分子作用相同的效应,则突变或敲除基因所编码的蛋白很有可能就是小分子作用的靶点。[5]实验方法准确性比较高,但是比较耗时费力,而且如果不能把小分子靶点锁定在一个较小的范围内的话,需要做大量的筛选工作,往往成本昂贵。而计算预测相对来说耗时比较少,但是当前算法的可靠性不如实验方法,通常理论计算得到的结果都需要实验来进一步验证。接下来将主要介绍靶点预测的一些计算方法。
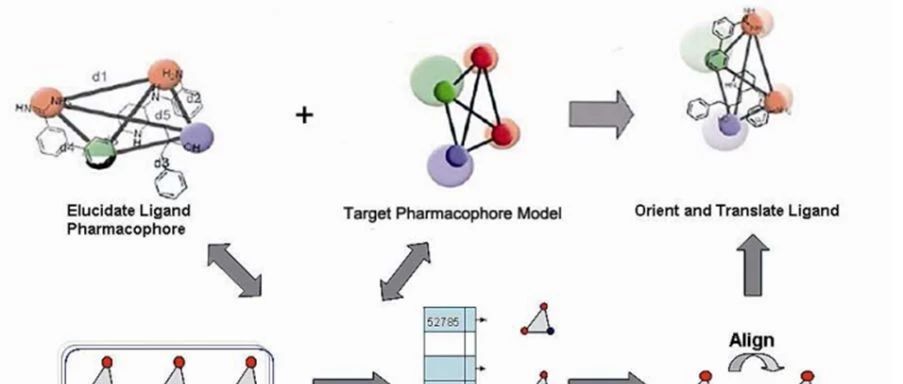
靶点理论预测方法中使用最多的是基于小分子配体结构的靶点发现,其核心理论就是结构相似的分子也会具有相似的生物学功能。[6]最简单的思路就是对于一个未知靶点的化合物,如果它与一个已知靶点的分子在结构上很相似,那它很有可能也可以作用于相同靶点。因此这个方法中最重要的一点就是化学结构的相似性搜索。
最常见的化学相似性检索方法有二维结构相似性和三维分子相似性检索。二维结构检索主要是采用分子指纹方法生成的描述符,分子指纹是指示化合物结构特征的一组数据,通常用0和1来表示,1表示分子中含有对应结构,0则表示没有,如图1A为PubChem分子指纹的一部分说明,如果分子中碳原子数量大于或等于2,则第9位为1,如果大于或等于4,则第9位、第10位都为1,以此类推。如果分子中含有碳氧双键结构(即C=O),则第420位为1,同样如果还含有C=S结构则421位也为1,否则为0。PubChem分子指纹一共预设了880种子结构特征,也就是说该分子指纹是一个880位的数组,包含的元素都是0或1。除了图中的子结构以外,PubChem分子指纹还对一些基团的数量有预定义,如第除了PubChem以外,常用的分子指纹还有MACCS、ECFP、Daylight fingerprints等分子指纹,它们的区别就在于子结构的定义以及编码的方式不同。计算得到分子指纹后就可以通过计算相似度(图1B)来比较两个分子之间的相似性。[7]以PubChem分子指纹为例,a为分子A中的子结构数量,b为分子B中的子结构数量,则c为分子A、B共有的结构数量,c的比例越大在一定程度上可以说明A、B分子越相似。因此这些相似度计算都是以c为判断标准,不同计算方法区别在于分母的不同(Hamming系数除外)。其中最常用的是Tanimoto系数,不同方法的比较在参考文献7中有详细阐述,本文不再赘述。
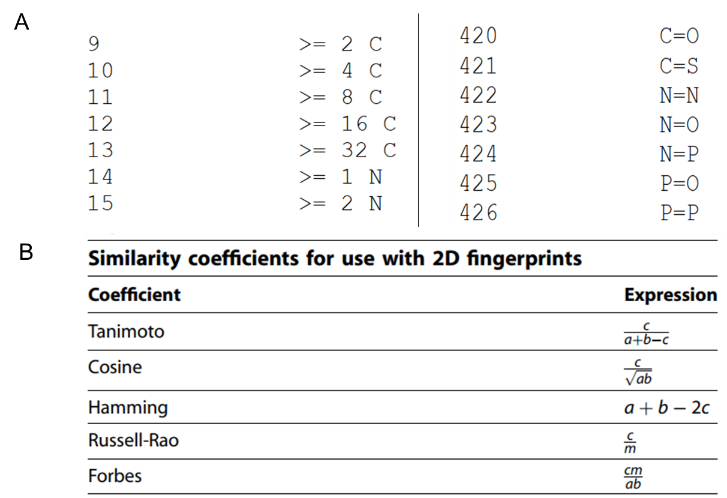
图1. A)PubChem分子指纹的部分子结构。B)分子指纹几种相似度计算方法,其中假设有两个分子A和B,则a表示分子A的分子指纹中1的数量,b表示分子B的分子指纹中1的数量,c表示A、B分子指纹中共同为1的数量,m表示分子指纹的总位数。
目前广泛使用的生物活性分子数据库如PDB、Binding DB、DrugBank、ChEMBL 等都带有以分子指纹为基础的化学结构相似性搜索功能,从而在某种程度上也可以提供简单的靶点预测。但是在这些数据库中一个分子的靶点可能有多个,对于用户来说设置不同相似度阈值也会得到不同数量的相似分子。因此有许多工具对这些数据库数据进行整合,并利用统计学等方法综合分子相似度和靶点分布情况给出一个合理的靶点预测结果。比如SEA(Similarity ensemble approach)[8]整合了ChEMBL和MDDR(MDL Drug Data Report)等数据库的化合物和靶点信息,利用Daylight分子指纹计算化合物的相似性,并将相似化合物的靶点进行聚类。用户输入化合物的SMILES结构式就可以在数据库中进行匹配,最终可以得到分子的潜在靶点列表(图2)。SuperPred[9]整合了SuperTarget, ChEMBL和BindingDB数据库,去掉了其中一些结合较弱(比如Ki、IC50值大于10μM)的化合物-蛋白相互作用,并且只保留了在Therapeutic Target Database(TTD)中有收录的靶点,也就是说所有的靶点都是和某些疾病相关的。SuperPred采取ECFP分子指纹计算结构相似性,支持化合物名称,SMILES以及用户自定义结构的查询,同样也可以得到预测靶点的相关信息(图3)。
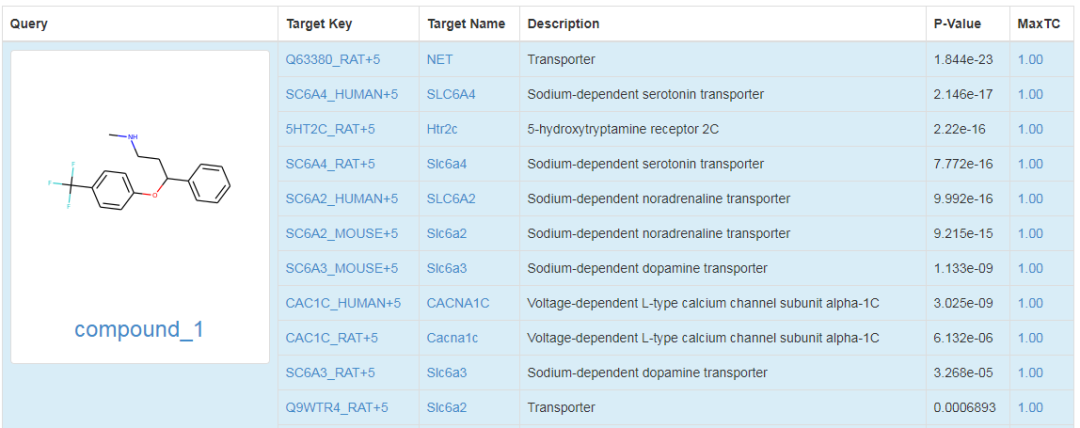
图2. SEA的搜索结果列表,包含了靶点的Unprot ID,显著性检验P值(P值越小成为靶点的可靠性越大),MaxTC表示数据库中和查询分子相似性最高的相关系数(Tanimoto Coefficient, 图1B)
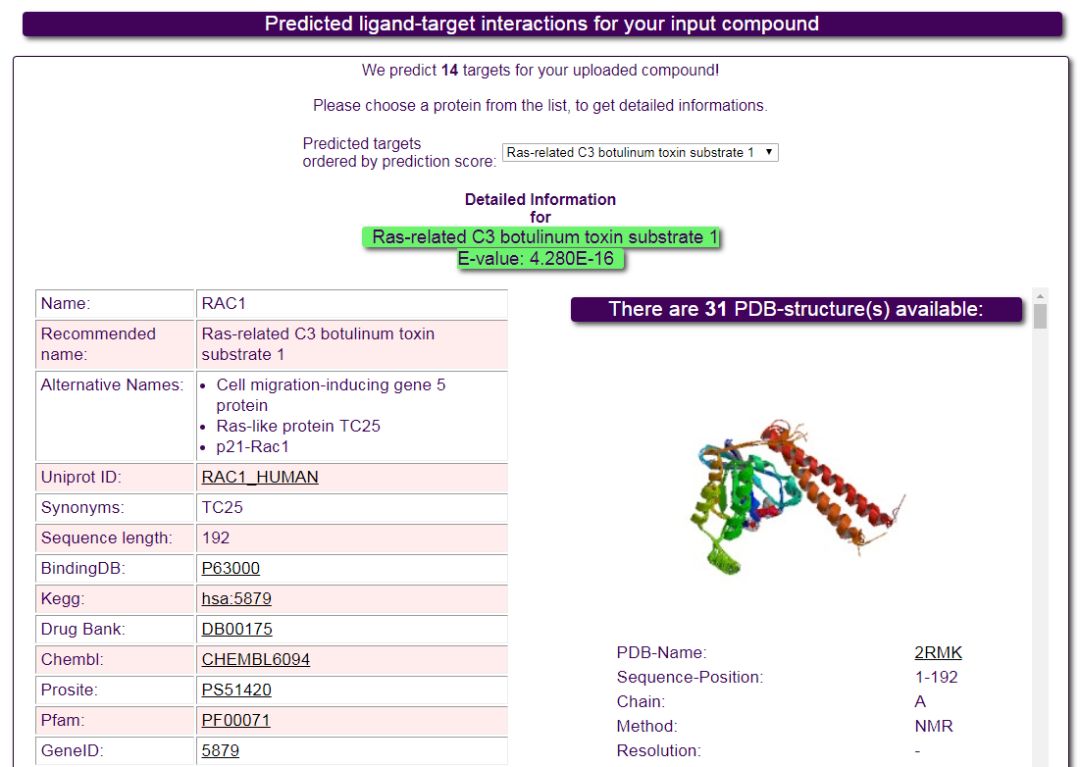
图3. SuperPred搜索结果列表,可以直接链接到Uniprot、BindingDB、DrugBank、PDB等数据库
除了二维分子指纹以外,三维分子相似性的计算也常用于靶点预测。ChemMapper[10]是一个基于SHAFTS (SHApe-FeaTure Similarity)[11]方法的靶点预测工具,主要是把三维分子的形状转化为数据进行比较。该方法首先定义了七种特征药效团,分别为疏水中心、正电中心、负电中心、氢键受体、氢键供体、芳香环以及金属离子螯合中心,这些药效团都分别对应了一系列的结构特征。对于待预测分子,先将分子中含有的上述药效团全部标记出来,每三个为一组在空间中组成一个三角形(图4),顶点代表不同类型的药效团。对于数据库中已知靶点的化合物做同样的操作,当待预测分子中的药效团三角形和已知化合物的药效团三角形相同(即两个三角形的顶点都是同样的三种药效团)时,称为一次匹配(match),然后单独对这两个三角形所包含的结构进行align,再计算他们的“FeatureScore”(图4中的FAB),FAB越大说明这两个分子相似性越高。
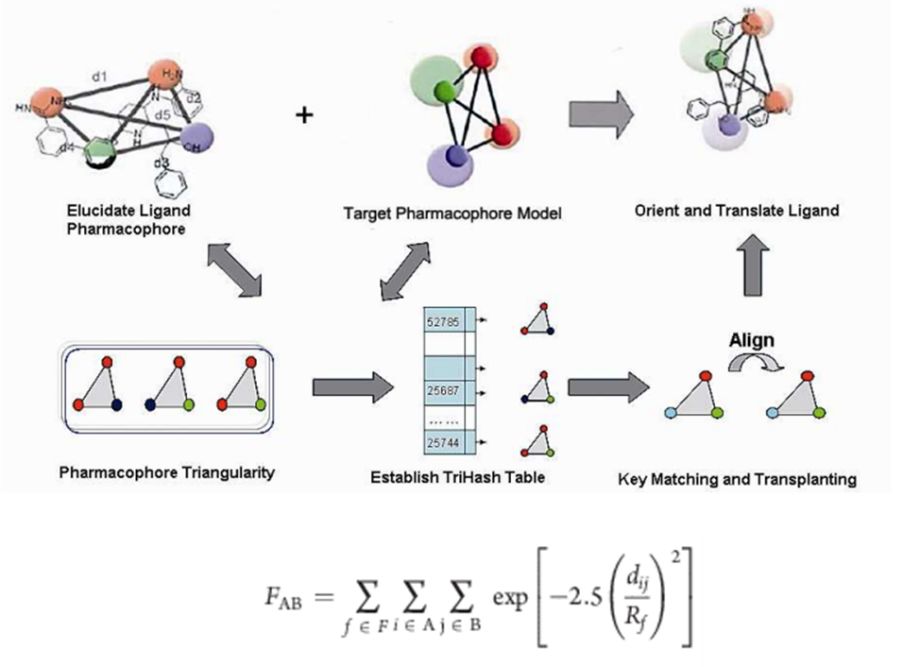
图4. 公式中i,j分别代表A分子和B分子中药效团所包含的原子,f为两分子产生的所有匹配F中的一种, dij为两原子距离,Rf是距离的可接受值,默认为0.8Å
靶点理论预测方法中还有一类基于受体结构特征的靶点预测,通常也指反向分子对接方法,即将小分子与一系列蛋白进行分子对接并进行打分,结合能力好的便认为是化合物的潜在靶点。这种方法的思路更接近于实验方法,但是将生化测试换成了分子对接,在速度上和实验相比要快很多,但是不如基于小分子配体的预测方法快速,而且这类方法很大程度上依赖于蛋白结构的准确性,该类方法的基本原理本文就不再详细阐述。表1列出了常用的开源在线靶点预测工具,可以发现靶点理论预测方法很大程度上依赖于一个完备的包含生物活性分子和靶点结构的数据库,其中ChEMBL、PDB是相对比较权威的数据库,其包含的信息也比较广泛。
表1. 常用的开源在线靶点预测工具
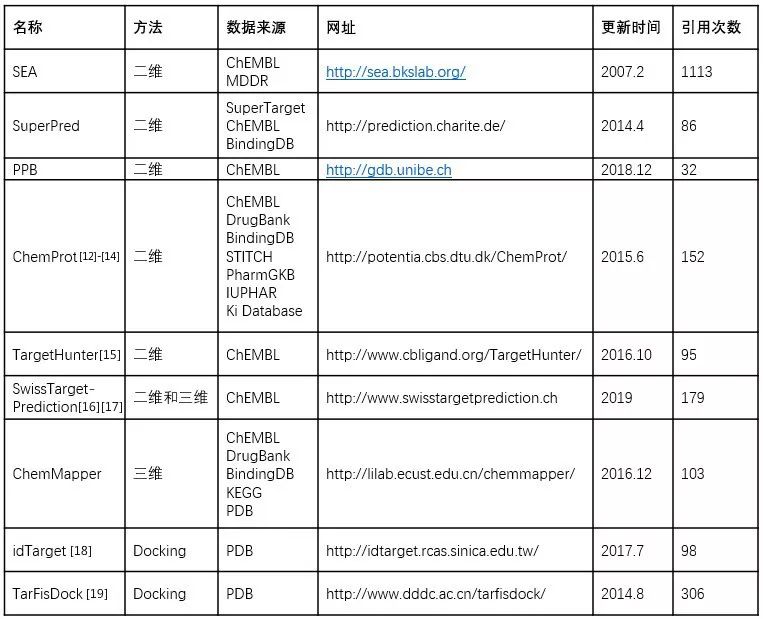
引用次数来源于Google scholar, 截至2019.8.26
随着生物活性数据以及化合物数据的增加,数据挖掘的方法也越来越多地应用于靶点预测,其中机器学习是最常见的方法。Polypharmacology Browser(PPB)[20][21]也是利用二维分子指纹相似性进行靶点预测的方法,在1.0版本中PPB融合了6种不同类型的分子指纹来进行分子相似性评估。而在2.0版本中,还是先将待预测分子与数据库中已知靶点分子用分子指纹进行相似性计算,然后把分子指纹作为特征,靶点作为分类标签,将相似性最高的前2000个分子应用于朴素贝叶斯模型进行训练分类,训练后的模型再以待预测分子的分子指纹为输入特征,输出分类进而得到靶点预测结果。
总的来说,化合物靶点理论预测方法已有不少但预测精度、广度往往还需要不断完善,且各类方法各有优劣。二维分子指纹的方法着重于分子化学结构的相似性,而三维方法更多的是着重于药效团的相似性。理论上说三维方法会比二维方法更加准确,因为化合物与靶点的结合需要考虑三维空间结构的匹配,而且根据药效团模型的理论,具有相同药效团的两个化合物在三维方法比较上是相似的但二维分子指纹的差异可能非常大。有文献指出在主靶点(化合物结合的主要靶点,治疗作用)预测上三维方法的优势不大,而对于脱靶效应(化合物结合的其他靶点,副作用)的预测上三维的方法要明显好于二维。[22]但是根据文献的报道,利用不同分子指纹以及三维相似性等的方法在各自的应用实例中表现也都比较好,不过对于大量用户提供的大量待预测分子是否都能很好地做出预测仍然有待检验。而且随着数据量的增加,数据的冗余和无用数据也在增加,因此对数据的筛选和清洗也是靶点预测需要考虑的地方。对于用户来说,如果条件允许,尽可能多地尝试不同预测工具,将得到的结果汇总后,再经人工判断有选择地对靶点进行实验验证,往往是当前比较切实可行的一个办法。
参考文献:
1. Gfeller, D.; Michielin, O.; Zoete, V., Shaping the Interaction Landscape of Bioactive Molecules. Bioinformatics 2013, 29, 3073-3079.
2. Sam, E.; Athri, P., Web-Based Drug Repurposing Tools: A Survey. Brief Bioinform 2017, 20, 299-316.
3. Forouzesh, A.; Samadi Foroushani, S.; Forouzesh, F.; Zand, E., Reliable Target Prediction of Bioactive Molecules Based on Chemical Similarity without Employing Statistical Methods. Front Pharmacol 2019, 10, 835.
4. Burdine, L.; Kodadek, T., Target Identification in Chemical Genetics: The (Often) Missing Link. Chem. Biol. 2004, 11, 593-597.
5. Zheng, X. S.; Chan, T.-F.; Zhou, H. H., Genetic and Genomic Approaches to Identify and Study the Targets of Bioactive Small Molecules. Chem. Biol. 2004, 11, 609-618.
6. Willett, P.; Barnard, J. M.; Downs, G. M., Chemical Similarity Searching. Journal of Chemical Information and Computer Sciences 1998, 38, 983-996.
7. Willett, P., Similarity-Based Virtual Screening Using 2d Fingerprints. Drug Discov Today 2006, 11, 1046-53.
8. Keiser, M. J.; Roth, B. L.; Armbruster, B. N.; Ernsberger, P.; Irwin, J. J.; Shoichet, B. K., Relating Protein Pharmacology by Ligand Chemistry. Nat. Biotechnol. 2007, 25, 197-206.
9. Nickel, J.; Gohlke, B.-O.; Erehman, J.; Banerjee, P.; Rong, W. W.; Goede, A.; Dunkel, M.; Preissner, R., Superpred: Update on Drug Classification and Target Prediction. Nucleic Acids Res. 2014, 42, W26-W31.
10. Gong, J.; Cai, C.; Liu, X.; Ku, X.; Jiang, H.; Gao, D.; Li, H., Chemmapper: A Versatile Web Server for Exploring Pharmacology and Chemical Structure Association Based on Molecular 3d Similarity Method. Bioinformatics 2013, 29, 1827-9.
11. Liu, X.; Jiang, H.; Li, H., Shafts: A Hybrid Approach for 3d Molecular Similarity Calculation. 1. Method and Assessment of Virtual Screening. J. Chem. Inf. Model. 2011, 51, 2372-2385.
12. Taboureau, O.; Nielsen, S. K.; Audouze, K.; Weinhold, N.; Edsgärd, D.; Roque, F. S.; Kouskoumvekaki, I.; Bora, A.; Curpan, R.; Jensen, T. S.; Brunak, S.; Oprea, T. I., Chemprot: A Disease Chemical Biology Database. Nucleic Acids Res. 2010, 39, D367-D372.
13. Kim Kjærulff, S.; Wich, L.; Kringelum, J.; Jacobsen, U. P.; Kouskoumvekaki, I.; Audouze, K.; Lund, O.; Brunak, S.; Oprea, T. I.; Taboureau, O., Chemprot-2.0: Visual Navigation in a Disease Chemical Biology Database. Nucleic Acids Res. 2012, 41, D464-D469.
14. Kringelum, J.; Kjaerulff, S. K.; Brunak, S.; Lund, O.; Oprea, T. I.; Taboureau, O., Chemprot-3.0: A Global Chemical Biology Diseases Mapping. Database 2016, 2016.
15. Wang, L.; Ma, C.; Wipf, P.; Liu, H.; Su, W.; Xie, X.-Q., Targethunter: An in Silico Target Identification Tool for Predicting Therapeutic Potential of Small Organic Molecules Based on Chemogenomic Database. The AAPS Journal 2013, 15, 395-406.
16. Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V., Swisstargetprediction: A Web Server for Target Prediction of Bioactive Small Molecules. Nucleic Acids Res. 2014, 42, W32-W38.
17. Daina, A.; Michielin, O.; Zoete, V., Swisstargetprediction: Updated Data and New Features for Efficient Prediction of Protein Targets of Small Molecules. Nucleic Acids Res. 2019, 47, W357-W364.
18. Wang, J.-C.; Chu, P.-Y.; Chen, C.-M.; Lin, J.-H., Idtarget: A Web Server for Identifying Protein Targets of Small Chemical Molecules with Robust Scoring Functions and a Divide-and-Conquer Docking Approach. Nucleic Acids Res. 2012, 40, W393-W399.
19. Li, H.; Gao, Z.; Kang, L.; Zhang, H.; Yang, K.; Yu, K.; Luo, X.; Zhu, W.; Chen, K.; Shen, J.; Wang, X.; Jiang, H., Tarfisdock: A Web Server for Identifying Drug Targets with Docking Approach. Nucleic Acids Res. 2006, 34, W219-W224.
20. Awale, M.; Reymond, J.-L., The Polypharmacology Browser: A Web-Based Multi-Fingerprint Target Prediction Tool Using Chembl Bioactivity Data. J. Cheminf. 2017, 9, 11.
21. Awale, M.; Reymond, J. L., Polypharmacology Browser Ppb2: Target Prediction Combining Nearest Neighbors with Machine Learning. J. Chem. Inf. Model. 2019, 59, 10-17.
22. Yera, E. R.; Cleves, A. E.; Jain, A. N., Chemical Structural Novelty: On-Targets and Off-Targets. J. Med. Chem. 2011, 54, 6771-6785.