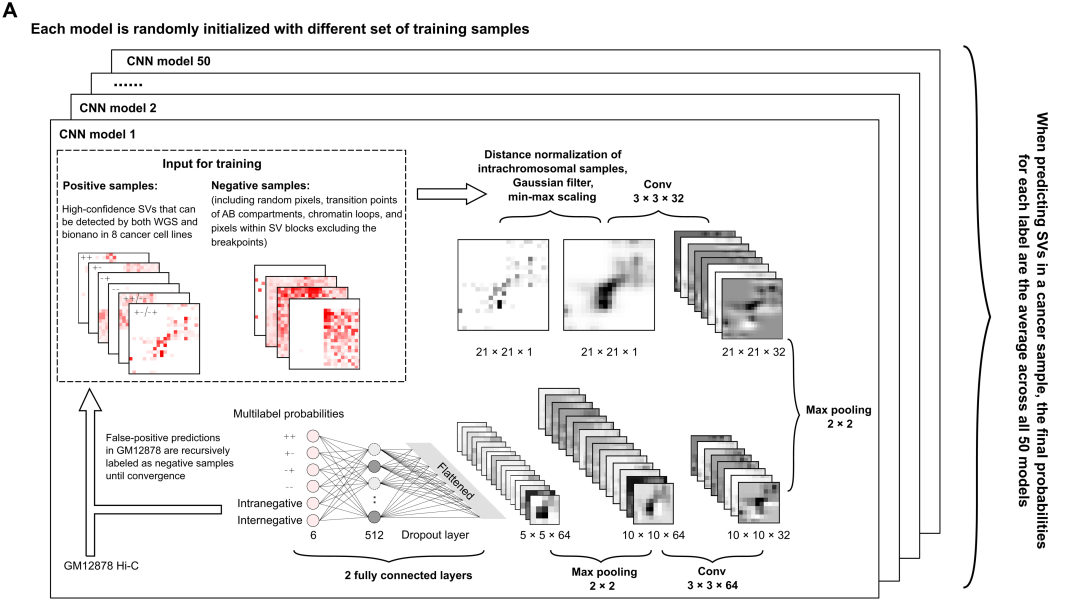

EagleC是一个结合了深度学习和集成学习策略的框架,能以高分辨率预测全范围的结构变异。
背景介绍
结构变异(SVs),包括缺失、倒置、重复和易位等,可通过多种机制直接导致肿瘤和其他疾病的发生。最近,研究表明,SVs可以将远端增强子带到原癌基因附近,并通过一种被称为增强子劫持的机制,导致致癌基因表达上调。复发性SVs的发现,大大提高了人们对肿瘤发生的认识,有利于有效的靶向治疗。
尽管它们很重要,但全基因组检测SVs仍然是一个具有挑战性的问题。传统上,核型分析一直是临床检测各种遗传病的主要方法,本质上是一种低吞吐量和低分辨率的方法。另外,基因芯片已被用于识别遗传物质的得失,但它在检测拷贝数中性事件如倒置和平衡易位方面有局限性。近年来,短读全基因组测序(short-read全基因组测序,WGS),因其高分辨率、高通量和简单性而被广泛用于识别各种基因组变异。但由于短读的可映射性问题,使用WGS很难检测到重复性区域的SVs。
最近,研究者及其他团队发现了Hi-C,一种最初被用于研究三维基因组结构的技术也可以用于系统的SV检测,基因组覆盖率低至1×。到目前为止,人们已经提出了三种利用Hi-C数据预测SVs的方法,但均有其局限性因此不是最优的。HiCtrans和HiNT-TL无法预测染色体内SVs,而Hi-C breakfinder只能检测到大小为>1 Mb的大的染色体内SVs。
主要内容
美国西北大学的岳峰等人开发出的EagleC可以独特地捕获一组全基因组测序或纳米孔缺失的融合基因,在精确度和召回率方面都优于现有方法。此外,EagleC还能在其他染色质相互作用平台上有效捕获SVs,如HiChIP、染色质相互作用分析与成对末端标签测序(ChIA-PET),并捕获Hi-C。研究者将EagleC应用于100多个癌症细胞系和原发肿瘤,并识别出一组有价值的高质量SVs。最后,研究者证明EagleC可以应用于单细胞Hi-C,并用于研究原发性肿瘤中SV的异质性。
EagleC框架概述
图1A描述了EagleC框架的总体设计。正训练样本被定义为围绕一组高置信度SVs的Hi-C接触矩阵,这些SVs通过WGS和光学图谱在8个癌细胞系(A549、Caki2、K562、LNCaP、NCI-H460、PANC-1、SK-N-MC和T47D)中被检测到。此外,为了使模型能够区分真实的SV信号和正常3D基因组特征诱导的假阳性信号,研究者从正常细胞系GM12878的Hi-C图谱中,采样了数量相近的染色体内和染色体间亚基,并分别标记为内负性和间负性。此外,还包括来自癌症Hi-C数据的矩阵,它们位于SV块中但不与断点重叠,是额外的负数据集。

图1. EagleC根据染色质相互作用数据预测了全范围的高分辨率SVs。图片来自Sci. Adv.
研究者使用训练样本的下采样版本,训练了一系列针对不同测序深度优化的EagleC模型。为了研究EagleC的性能,研究者预测了训练过程中未使用的其他癌症Hi-C数据集中的SVs(文中默认所有SVs分辨率均为5kb)。EagleC成功预测了不同类型的SVs,包括断点距离小于1 Mb甚至100 kb的短程SVs(图1,B-D)、大的染色体内SVs(图1E)、互易的染色体间易位(图1F)和非互易的染色体间易位(图1G)。
EagleC在Hi-C图上检测SVs中优于现有的方法
研究者首先直观地检查了预测结果,发现几乎所有具有异常高相互作用频率的块都被预测为SVs,这表明框架具有很高的灵敏度(图2A)。在许多情况下,虽然EagleC和Hi-C breakfinder预测了相同的SV块,但预测断点的确切坐标不同,EagleC预测断点更有可能被WGS验证(图2A,区域“A”,“C”,“D”和“E”)。此外,EagleC在5kb分辨率下预测的断点比Hi-C breakfinder预测的更精确,Hi-C breakfinder通常是100kb分辨率。

图2. EagleC在精确度和召回率上的优越表现。图片来自Sci. Adv.
然后,研究者对两种目前唯一可以识别染色体内SVs的方法—EagleC和Hi-C breakfinder进行了更深入的比较。值得注意的是,EagleC在BT-474、HCC1954和MCF7中检测到的SVs(包括染色体间易位和染色体内SVs)分别是Hi-C breakfinder的2.4倍(244/100)、2.6倍(410/157)和4.8倍(244/51)(图2B)。同时,EagleC在这些细胞系中取得了明显高于Hi-C breakfinder的精度率。
在BT-474中,24.2%的EagleC预测的SVs与59.0%的Hi-C breakfinder预测相匹配。在EagleC特有的185个SVs中,83.2%可以通过WGS或纳米孔进行验证,相比之下,Hi-C breakfinder特有SVs的验证率为2.4% (图2C)。
接下来,研究者将分析扩展到另外26个具有Hi-C和WGS数据的癌症细胞系或患者样本。再次观察到,与Hi-C breakfinder相比,EagleC在所有26个癌症样本中取得了显著更高的召回率和准确率(图2D-F)。由于算法本身的局限性,Hi-C breakfinder只能检测到大于1 Mb的大的染色体内SVs。但是,如图2G所示,EagleC预测的39.5%的染色体内SVs是短距离SVs,最小为35 kb。令人惊讶的是,尽管人们认为很难将这一范围的SVs与其他Hi-C接触模式区分开来,但它们的预测精度甚至高于远程SVs和易位(图2H)。
EagleC检测癌症中新的融合基因
如图3A所示,EagleC在MCF7中检测到了ATXN7和BCAS3基因内部的断点,而arriba软件也预测了这两个基因的融合(图3A,右)。研究者在图3中展示了另外两个这样的例子,表明由于EagleC的高分辨率特性,它可以独特地预测WGS和纳米孔缺失的融合基因。另外,与未进行融合的非恶性细胞系相比,参与这些融合事件的基因在癌细胞中显著过表达(图3D)。

图3. EagleC唯一检测到的融合基因在癌细胞中过表达。图片来自Sci. Adv.
EagleC可以使用其他基于3C的技术准确地预测SVs
研究者直接将Hi-C数据训练的EagleC模型,应用于CTCF ChIA-PET和Pol2 ChIA-PET。总的来说,EagleC在Hi-C、CTCF ChIA-PET和Pol2 ChIA-PET中预测了类似数量的SVs,并且三个数据集之间有很大的重叠(图4A-B)。例如,EagleC在CTCF ChIA-PET中预测了226个SVs,其中66.4%在Hi-C中也预测到了。同样,Pol2 ChIA-PET中预测的62.8%(196个中的123个)SVs与Hi-C预测的50.4%(244个中的123个)相匹配。就精度而言,相比于Hi-C(73.8%),EagleC在两个ChIA-PET数据集中取得了相当的精度(CTCF,65.5%;和Pol2, 68.2%)(图4C)。此外,研究者观察到,在所有10个与WGS数据匹配的HiChIP/ChIA-PET数据集中,EagleC预测的SVs的召回率和准确率都显著高于Hi-C breakfinder(图4D-F)。

图4. EagleC可以准确预测HiChIP和ChIA-PET接触图上的SVs。图片来自Sci. Adv.
105例肿瘤标本中SVs的检测
如果在同一个样本中有多个数据集可用,将它们的结果结合起来可以形成更全面的SV注释集。研究者预测了所有样本中的5620个SVs,每个样本中的数量从2到410不等(图5A)。所有样本数据中,30.9%的预测SVs是短程SVs (<1 Mb),35.7%是长程SVs,33.4%是染色体间易位。
在超级酶尺度上,哺乳动物的基因组被组织成TADs。CTCF结合位点富集的TAD边界为适当的基因调控提供了一个绝缘的环境。与随机打乱的SV的预期分布相比,研究者发现SV的断点明显靠近TAD边界,这与之前的研究结果一致,即DNA拓扑异构酶II beta (TOP2B)介导的DNA双链断裂在染色质环的锚点富集(图5C)。总体而言,约10%的SVs发生在TAD边界之间,37.5%发生在TAD边界与TAD内区域之间,52.5%发生在TAD内区域之间(图5D)。此外,研究者发现癌症相关基因的转录起始位点(TSSs)在断点相关的TAD边界特异富集(图5E)。这表明基因组重排列破坏TAD边界,可能是致癌基因失调和肿瘤发生的重要机制。

图5. 105株癌细胞或患者样本中SVs的泛癌分析。图片来自Sci. Adv.
为了进一步探索研究者的SV注释的价值,研究者鉴定了不同样本中反复受到短程SV影响的基因。结果发现大部分缺失的基因是肿瘤抑制基因(图5F),如CDKN2A/2B、WWOX、CHFR和MSH2基因。另一方面,在重复区域内的很多基因都是癌基因(图5G),如MYC。CD44基因是癌症干细胞的常见生物标志物,编码一种参与肿瘤起始和进展的细胞表面糖蛋白。
EagleC预测单细胞中已知的染色体间易位
为了使EagleC在每个细胞接触信息有限的情况下适用于scHi-C,研究者将相同的8个癌细胞株和GM12878细胞的接触图谱降采样到可比较的测序深度,并在500 kb分辨率下重新训练模型。然后,研究者在HAP1和K562中发表的scHi-C数据集上测试了EagleC,这两种细胞都是慢性髓系白血病细胞系。HAP1细胞中9号染色体和22号染色体为互易位,而K562细胞中9号染色体和22号染色体为非互易位。HAP1数据集包含256个单细胞,每个细胞中位数为18793个接触,而K562数据集包含337个细胞,每个细胞中位数仅为3974个接触(图6A)。值得注意的是,即使使用这些极其稀疏的接触矩阵,EagleC也能够预测单细胞中已知的chr9-chr22易位(图6B-C)。

图6. EagleC可以准确预测HiChIP和ChIA-PET接触图上的SVs。图片来自Sci. Adv.
为了系统地研究准确预测单个细胞中SVs的接触数下限,研究者根据测序深度对所有256个HAP1细胞进行了排序,并通过汇聚到99个最深的单个细胞产生了一系列的接触矩阵(接触对从148635万到405万)(图6D)。正如预期的那样,预测的SVs数量随着细胞数量的增加而减少(图6E)。
结论总结
本文利用CNNs在图像识别和集成学习避免过拟合问题的优势开发的EagleC不仅能预测出唯一的近距离SVs,且与现有方法相比,大大提高了整体预测能力。本文证明了利用Hi-C检测融合基因的可行性,虽然目前的框架无法达到碱基对分辨率,但与RNA-seq相比,Hi-C在检测内含子内融合点方面具有独特的能力。此外,EagleC可以作为一个通用模型来预测SVs,使用其他基于3C的接触图,包括ChIA-PET,HiChIP/PLAC-Seq,capture Hi-C,甚至是scHi-C。
参考文献
Wang X, Luan Y, Yue F. EagleC: A deep-learning framework for detecting a full range of structural variations from bulk and single-cell contact maps[J]. Science Advances, 2022, 8(24): eabn9215.