先睹为快
靶点
胰高血糖素样肽1-受体(GLP-1R)
计算方法
分子对接,分子建模
计算软件
Schrödinger,MOE,Cubist
单位与作者信息
赛诺菲-安万特María Méndez,Hans Matter
发表杂志
Journal of Medicinal Chemistry
计算流程
将胰高血糖素样肽1-受体-肽激动剂复合物晶体结构(PDB code: 5NX2)导入Schrödinger的蛋白质准备模块进行准备,用LigPrep制备配体结构,并使用Glide XP将配体对接至GLP-1R模型中,Induced fit docking用于限制参考配体9的对接姿势,其对内核的弱约束力使吲哚部分趋于合理排列,给其余配体对接提供指导。ADME模型则由Corina生成规范3D几何坐标,基于MOE产生2D-QSAR。Cubist构建一个基于规则的决策树并用C5.0开发定量分类模型,对259种化合物进行性质预测。
背景

胰高血糖素样肽1-受体属于B类G蛋白偶联受体(GPCR),是治疗2型糖尿病(T2DM)的有效靶点。目前市面上被批准用于治疗T2DM的药物多是可注射的几种GLP-1R肽激动剂,在使用过程中普遍存在恶心和呕吐的现象,这激发了研究人员寻找肽变体或充当GLP-1R激动剂或正变构调节剂(PAMs)的小分子的热情。尤其是PAM,做成口服液可大大提高患者的便利性以及对最大治疗功效的依从性且PAM不会干扰内源肽水平,这是对抗完全激动剂的一大优势。目前采用两种方法来用小分子激活GLP-1R,寻找能够增强内源性配体GLP-1(7-36)NH2或活性低得多的降解产物GLP1(9-36)NH2的活性的直接激动剂或PAM。较早的GLP-1R小分子激动剂有非药性质,如具有高亲脂性和分子量,口服后血浆暴露量会降低。vTv Therapeutics,辉瑞及礼来针对GLP-1R的直接激动剂的研发中开发了共价和非共价的PAMs,但均对化合物如何与T2DM疾病模型进行关联及其体内药学信息知之甚少。基于GLP1(9-36)NH2的体内活性高于GLP-1(7-36)NH2的事实,作者提出了增强GLP1(9-36)NH2的活性会产生有价值的治疗方法的假设且用高通量筛选的方法对这一假设做出论证。
HTS发现GLP-1R的正向变构调节剂
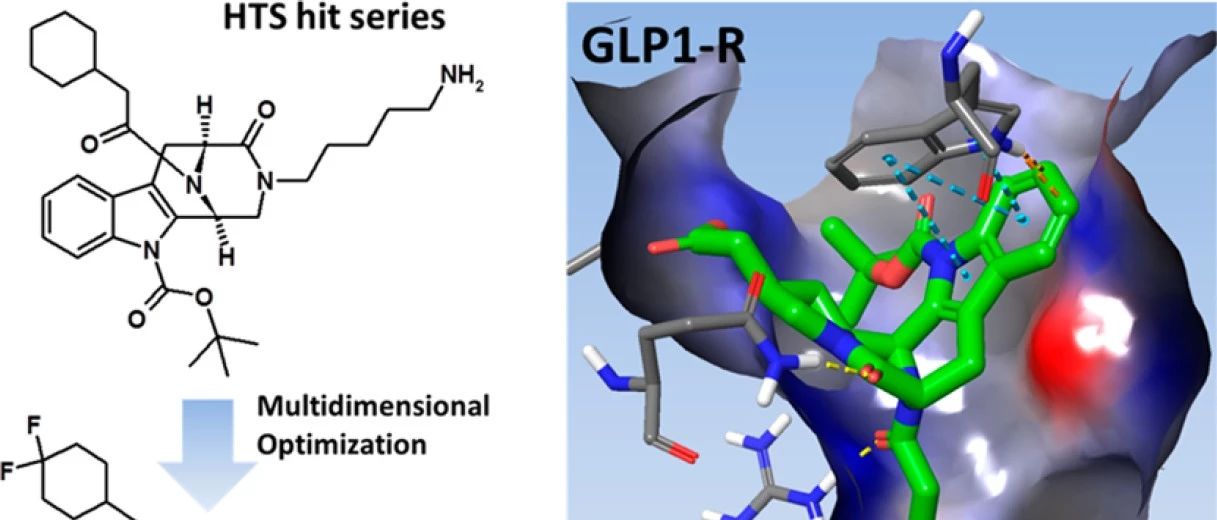
作者对赛诺菲的化合物进行了HTS鉴定,以确定内源性配体GLP1(9-36)NH2与GLP-1R相互作用的正向变构调节剂。该实验在过度表达的人GLP-1R的HEK293细胞系中使用HTRF cAMP分析完成的。GLP-1(7-36)NH2的EC50值大约是GLP1(9-36)NH2的10000倍。进行筛选分析时,所有化合物均在正构配体GLP1(9–36)NH2的EC20浓度下进行测试,以更好地评估推定的HTS命中强度。随后,测试了化合物在固定浓度为10和3μM的条件下改变内源性GLP1(9-36)NH2的EC 50值的能力,从而证明活性较低的内源性肽具有改善的生物学作用。一个基于3,4,5,6-tetrahydro-1H-1,5-epiminoazocino[4,5-b]吲哚骨架的化学系列在HTS分析中显示特别高的活性(化合物8,PAM EC50 600nM/100%有效),在同源小鼠受体测定中,生物学活性相似或略有改善(PAM EC50 300nM/100%)。这一化合物系列来源于使用Pictet-Spengler分子内环化技术,基于类天然产物的骨架合成的化学文库。该来源使研究人员能快速进行结构活性关系(SAR)评估。化合物8的高生物活性促使作者明白桥头碳原子的立体化学式高GLP-1R活性所必需的的结构,由此,对8的结构进行进一步优化。
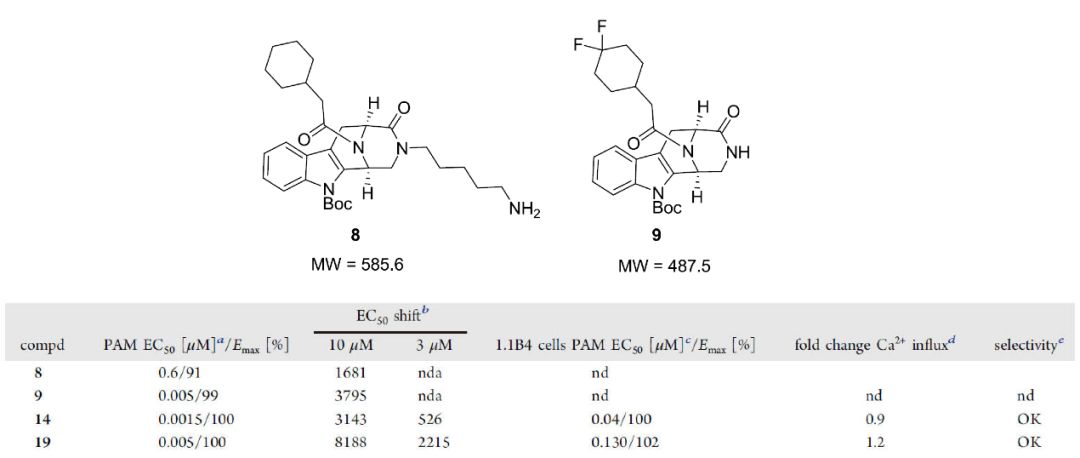
图1. 化合物8和9的结构及化合物8-19的体外数据汇总
图片来源JMC
正向变构调节剂的合成及其分子药理学研究
化合物19的合成采用略微修饰的合成方法。从Fmoc保护的色氨酸9a和胺结构单元15开始,通过HATU偶联获得中间体16。用Dess-Martin高碘烷氧化乙醇等一列系操作的得到了作为单一对映异构体的化合物19。14和19都是HTS命中的化合物8经进一步优化得到的类似物,二者在酰胺氮上均带有极性取代基。在体外测定中,两种化合物均显示出相似的效力(化合物14和化合物19的 EC50/Emax 分别为1.5 nM/100%,5 nM/100%),但是,19在移动GLP1(9-36)NH2的EC50值的主要实验中比化合物14表现优异(化合物19和14在10和3μM对EC50的改变分别为8188/2215和3143/526)。这是迄今为止报道的针对GLP-1R非共价PAM的最佳强化能力。化合物14和19均被证明是GLP1(9-36)NH2的选择性激动剂,在葡萄糖刺激的胰岛素(GSIS)分析中,化合物14显著改善了GLP1(9-36)NH2介导的葡萄糖刺激的胰岛素在大鼠胰岛中的分泌。在更高浓度的肽配体中,这种GSIS增强作用更加明显,但浓度过高时,14增强GLP1(9-36)NH2效果的能力消失,甚至变为抑制作用,而化合物19即便在高浓度(10μM)时,也能强烈刺激大鼠胰岛中的GSIS现象。且该实验在低葡萄糖浓度下不存在促胰岛素反应。
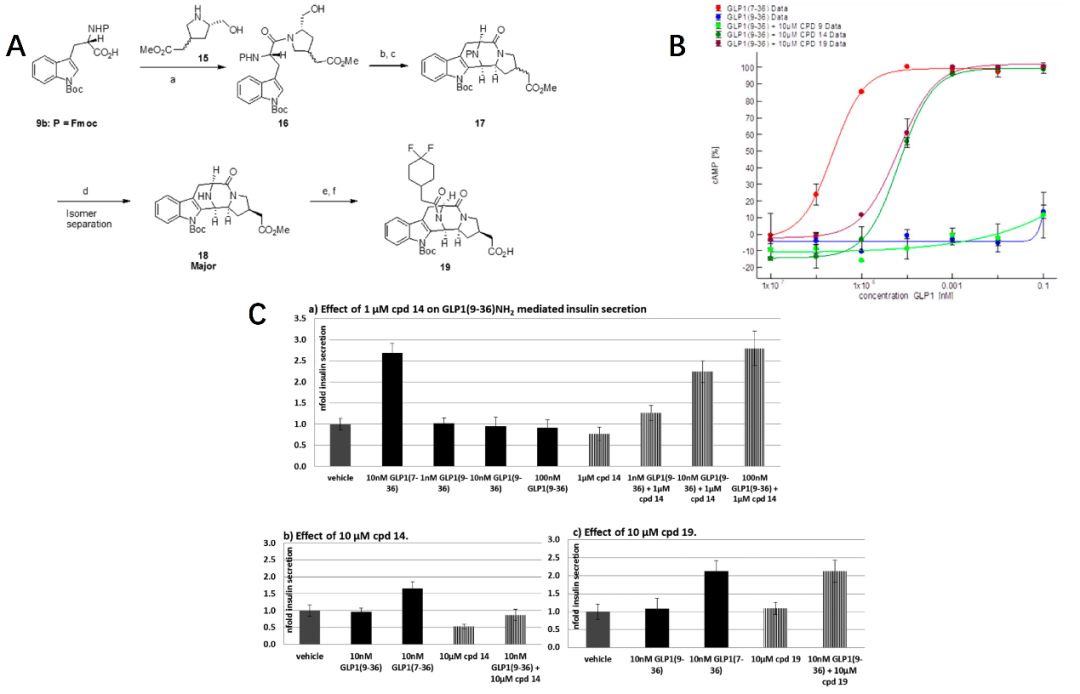
图2. 化合物19的合成路线(A), 14和19功能性测定的剂量反应曲线(B)和及GSIS分析(C)
图片来源JMC
GLP-1R中配体结合模式假说
使用处于活性构象中的肽激动剂与全长GLP-1R的X射线复合物结构(PDB code:5NX2),得出了3,4,5,6-tetrahydro-1 H -1,5-epiminoazocino[4,5-b]吲哚骨架的结合假设,由于缺乏共晶结构和GLP-1R突变体,这仅是一个可以说明作者进行配体优化的理由,不能排除该系列其他潜在结合方式。在对接分析中观察到文中系列化合物与正构口袋重叠的区域:(1) 配体在添加截短的肽GLP1(9-36)NH2时表现出正变构作用但在完整GLP-1(7–36)NH2存在下,氨酸His7-Ala8靠近变构位点,阻止了配体进入该区域; (2) 只有通过使ECD与截短的GLP-1肽衍生物和7TM结构域接触,才能激活GLP-1R; (3)结合位点假说允许我们在空间上适应作者的配体系列。对于化合物9,诱导契合对接得出了与SAR一致的假设。叔丁基可能位于Leu388旁边,环外配体酰胺氧可能与对GLP-1结合亲和力至关重要的Arg190相互作用。二氟环己基可能位于Tyr241附近的疏水口袋中。哌嗪酮酰胺可能与Gln234和Met233相互作用,后者是肽结合的重要残基。吲哚可能位于靠近Trp306和Arg380的上口袋区域。化合物14和19与GLP-1R的结合模式与化合物9类似,其中,19的羧酸可能与Lys197的侧链接触。
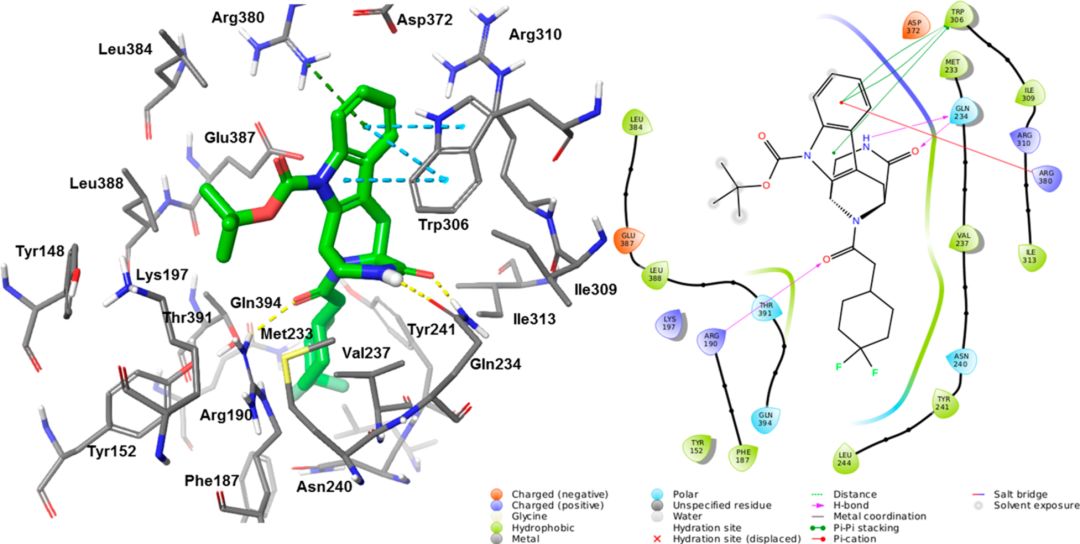
图3.通过诱导契合对接,化合物9与GLP-1R的X射线结构的结合模式
图片来源JMC
化合物19的体外ADME和体内药代动力学分析
基于化合物19在体外药理实验中令人鼓舞的表现,作者评估了其ADME性质,以评估其在体内研究中作为工具化合物对概念验证(PoC)的潜力。化合物19在人和小鼠肝微粒体中代谢稳定,并且在Caco-2 TC7细胞转运实验中表现出高细胞通透性。此外,该化合物不抑制CYP3A4,显示出良好的水溶性,并且在包括GSH和小鼠血浆在内的不同条件下化学稳定。在小鼠肝细胞中,19的中间清除率为0.145(mL/h)/106个细胞,相当于肝血流量的20%。作者利用过表达人类有机阴离子转运蛋白hOATP1B1和hOATP1B3的HEK293细胞进行了转运蛋白研究,用根据内部化合物和数据构建的2D-QSAR模型成功预测了19与OATP1B1和OATP1B3的相互作用,并且19也被分类为P-gp的底物。
在利福平的存在下将化合物19应用在ob/ob小鼠中的概念验证研究,这是公认的T2DM疾病模型。在所有使用的模型中,在没有利福平和/或GLP1(9–36)NH2的情况下,单独的化合物19均未显示对血糖浓度的任何影响。在肽GLP1(9–36)NH2的所有测试剂量下,化合物19在oGTT期间均引起血糖浓度的显着剂量依赖性降低。这正好支持了作者的假设,即该正变构调节剂19的药理作用是其直接与GLP-1受体产生相互作用。
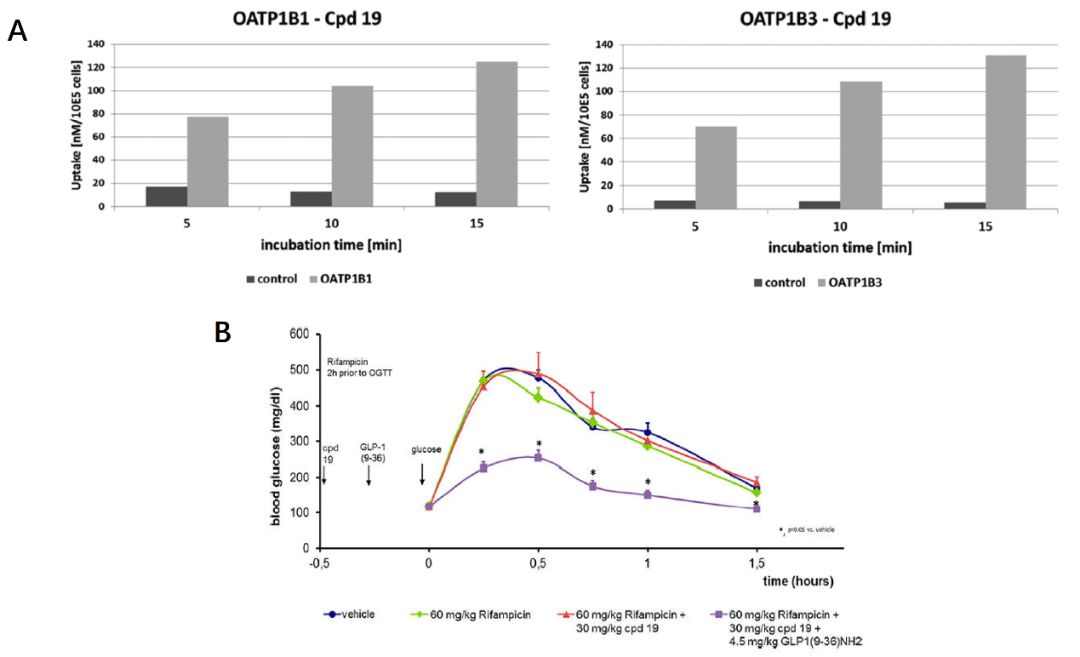
图4. 化合物19化合物19被鉴定为hOATP1B1和hOATP1B3转运蛋白的底物(A)和其体内药代动力学分析(B)
图片来源JMC
总结
本文利用高通量筛选和分子建模以及体内外实验验证的方法,开发了一类含有新骨架的胰高血糖素样肽-1受体的有效正向变构调节剂,并对这系列化合物进行结构优化,合成和药理学评估。其中,化合物19在各项功能测定中均显示出高活性和选择性,该PAM能将活性显著降低的代谢物GLP1(9–36)NH2转变为有效的配体,该配体能够以与活性GLP-1肽本身相似的方式激活GLP-1R,为2型糖尿病的治疗寻找到一类新颖的GLP-1R小分子激动剂的骨架。
参考文献
Mendez M, Matter H, Defossa E, et al. Design, Synthesis, and Pharmacological Evaluation of Potent Positive Allosteric Modulators of the Glucagon-like Peptide-1 Receptor (GLP-1R). J Med Chem 2019.