

背景介绍
自从人们认识到药物结合动力学速率可能是体内药物功效的关键决定因素,对开发计算方法的兴趣就日渐增加,以期能估计出药物-靶标解离速率常数 (koff) 或停留时间 (τ= 1 /koff)。业界已有的能预测动力学速率并推导出定量结构-动力学关系 (QSKRs)的方法大多依赖晶体结构而每个配体-蛋白复合物只能被视为具有单一结构。海德堡理论研究所的Wade教授团队提出了比较结合能量(COMBINE)分析的改进方法,在COMBINE分析中引入了使用多个结构来描述每个配体-蛋白质复合物的选项,并将其应用于研究蛋白质结合口袋柔性对p38丝裂原活化蛋白 (MAP) 激酶抑制剂的解离速率常数 (koff) 推导的影响。通过对从分子动力学模拟获得的蛋白质构型进行对接,为每个配体-蛋白质复合物获得了多个结构,然后通过最小二乘回归获得从配体-蛋白质复合物的能量最小化结构确定的配体-蛋白质相互作用能的比例系数,进而用于计算koff值。本文研究旨在扩展COMBIE分析方法,以允许使用一组结构来代表一个配体-蛋白质复合物,从而将结构柔性的影响纳入结合参数的预测中。作者建立了两个COMBINE分析模型来预测koff值:一个使用能量最小化的晶体结构来表示每个配体-蛋白质复合物,另一个使用来自MD模拟的用于集成对接的10个结构来表示每个配体-蛋白质复合物。通过选择p38丝裂原活化蛋白 (MAP) 激酶作为模型系统,由于其实验koff值的范围和晶体结构的可用性,证明了COMBINE分析是一种很有前景的方法,可以指导设计一些能以改进的结合动力学结合柔性蛋白质的化合物。
主要内容
1. 使用一个晶体结构代表一种配体-蛋白复合物的COMBINE分析
为了评价COMBINE分析中纳入蛋白质柔性的有用性,作者使用来自X射线晶体学的一个能量最小化结构来表示每个配体-蛋白质复合物并获得相互作用能。作者计算了训练集中22种 p38 MAP 激酶抑制剂中每一种抑制剂与348个残基的348个静电和 348个范德华相互作用能。然后选择标准偏差大于指定截断值(0.25 kcal/mol)的相互作用能,以去除可能对koff值差异没有贡献的能量项,最终选择了36个范德华相互作用能和14个静电相互作用能。接下来,使用 PLS回归确定这50个相互作用能量对于不同数量的潜在变量的权重,以将相互作用能量与实验 log koff 值相关联(图1A)。选择具有两个潜在变量的模型是因为它在使用留一法(leave-one-out)进行交叉验证时呈现最高的Q2值 0.75。此外,对于所使用的不同交叉验证方法,Q2、AAEv 和RMEv 值相似,表明具有两个潜在变量的模型是一致的。当从留一法转向留二法和留三法时,Q2的下降是预料之中的,因为后者的训练集中使用的结构较少。最终该模型的决定系数R2tr值为 0.86,表明它可以成功拟合训练集(图1B)。测试集的决定系数 (R2te) 为0.79(图1B),表明该模型具有较高的预测能力。权重最高的相互作用能是抑制剂与Lys53、Arg70、Glu71和Leu167残基的范德华相互作用能,以及与结合位点周围的Lys53和Glu71残基的静电相互作用能(图 1A、C)。与Lys53静电相互作用的权重为负,这意味着与该残基的有利相互作用将导致高koff值。另一方面,与Glu71的静电相互作用和大多数范德华相互作用的权重为正,表明与这些残基的有利相互作用将导致低koff值。

图 1. COMBINE分析模型计算 p38 MAP激酶抑制剂的koff值。图片来源于JCIM
2. 使用来自MD的多个结构来表示一种配体-蛋白质复合物的COMBINE分析
从p38 MAP MD模拟轨迹中挑选了10个构象,然后将33种p38 MAP激酶抑制剂与10个构象依次进行对接,总共获得330种配体-蛋白质复合物。对于大多数配体,从集成对接中获得的配体构象相似,并且在COMBINE模型中使用的复合物中的配体构象具有较低的RMSD值。将这些不同的结合构象同样进行能量计算和回归分析(图1B), 结果不仅包括了使用单一结构确定的一些主要贡献,例如与Lys53 和Glu71的静电相互作用以及与Glu71和Leu167 的范德华相互作用,而且还发现了与DFG基序中残基 Arg67和Asp168 相互作用的突出贡献,它们在 MD 模拟期间更接近结合位点,并且与配体的长时间驻留密切相关(图2)。
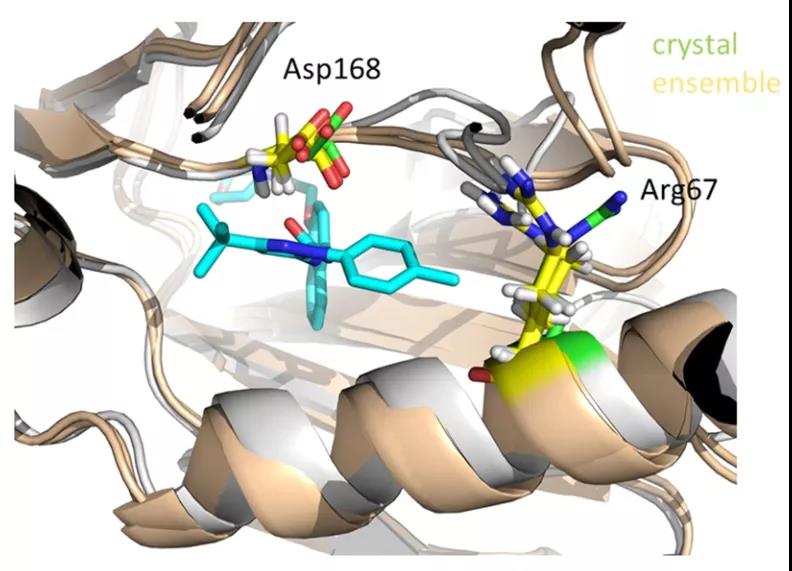
图2. 在COMBINE分析中使用多个结构进而考虑蛋白质和配体的柔性有助于突出与残基Arg67和Asp168相互作用的贡献。图片来自JCIM
3. 使用单个或多个结构的模型比较
使用一个或多个结构从COMBINE分析中获得了类似的R2tr 值(分别为0.86和0.83),表明两种模型基本上都可以很好地拟合训练集。两种模型还可以重现由配体13(训练集)和3、14(测试集)组成的小配体系列的排名,它们仅相差一个基团(图3)。然而,使用一个结构的模型的整体R2te值较高,表明在具有一个结构的模型中选择的相互作用能组具有更高的预测能力。在多个结构模型中,选择的相互作用能的预测能力较低的可能原因包括较差的信噪比、重要蛋白质构象的采样不足以及对接获得的配体位置不佳等。
尽管与具有单一结构的模型相比计算时间有所增加,但对一个复合物使用多结构的 COMBINE 分析在蛋白柔性不可忽视的情况下非常有用。在这里,p38 MAP 激酶中靠近结合位点的区域在所有晶体结构中都没有解析出来,这表明该区域是高度灵活的。使用多结构COMBINE分析有助于揭示更多参与 koff 值调节的残基。多结构COMBINE也可应用于配体具有多种结合模式或蛋白口袋内残基具有多种构象或旋转异构体的情况。
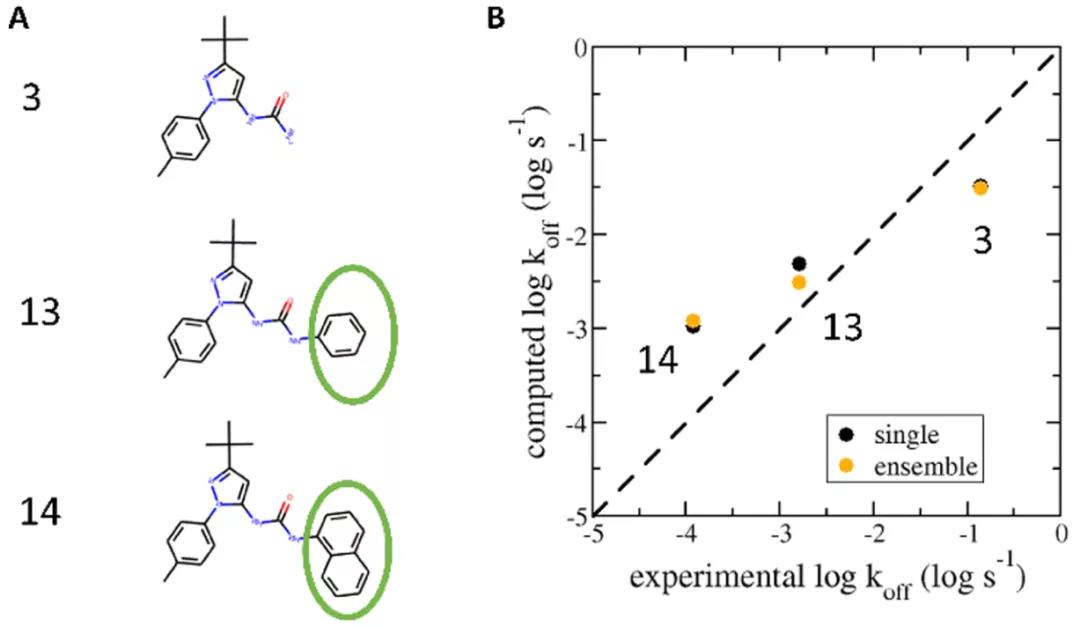
图3. 通过COMBINE分析计算的p38 MAP激酶抑制剂koff值可以重现由配体 13(训练集)和3和14(测试集)的排名。图片来自JCIM
小结
本文提出了一种将蛋白质和配体柔性纳入COMBINE分析的方法,该方法使用来自描述一个配体-蛋白质复合物的多个结构来预测koff值。作者获得了具有良好预测能力的p38 MAP激酶抑制剂的koff值模型,并确定了导致结合动力学变化的关键配体-蛋白质相互作用。此外,配体-蛋白质柔性的加入有助于突出与长停留时间相关的更多残基,这些特定的相互作用能有助于深入了解慢速解离抑制剂和快速解离抑制剂的作用机制。
参考文献
Nunes-Alves A, Ormersbach F, Wade RC. Prediction of the Drug-Target Binding Kinetics for Flexible Proteins by Comparative Binding Energy Analysis. J Chem Inf Model. 2021 Jul 26.