

背景介绍
在有机合成领域,化学家不仅要知道合成分子的步骤和顺序,还要知道反应条件,并希望每步反应的产率足够高。对于化学家来说,理解高维反应数据的相互作用并抽象出新的规律是一个难题。本文介绍了如何采集数据,以及如何使用无监督模型减少生成新反应数据集时的偏差,尤其关注模型选择、比较、训练以及化学可解释性。
首先,作者在单个Ni 催化的铃木交叉偶联反应的小型数据集中评估了 16 个膦配体,使用线性回归模型预测产率。发现模型的复杂度受到数据量的约束,于是尝试筛选了反应直接相关的特征。然后用更大的数据集,探索更复杂的模型。作者用这些数据集研究了各种 ML模型,并测试了这些模型的泛化能力。
主要内容
1. 实验数据
实验的数据既可以从公开的反应数据库中挖掘也可以从头生成。前者作者通过与MIT、辉瑞和默克等大型机构合作获得海量信息,后者依赖化学家的经验来为给定反应选择底物和条件。如果化学家提供的条件在化学空间中与其他条件差异太大,模型参数就会受到较大的影响,使用无监督机器学习的数据集设计系统方法,例如基于特征的聚类,是一个潜在的解决方案。
2. 特征工程
分子特征可以大致分为非学习和学习两种表示方法。前者从构建的描述符和预测目标入手建立模型。但是这种表示方法仅适用于大数据集。我们使用密度泛函理论 (DFT) 获取底物、催化剂和试剂的特征。这种方法能降低计算成本,但是会牺牲中小型数据集的可解释性和适用性。非学习特征依赖化学知识或先验机制。例如,我们在镍催化的铃木偶联反应中最初筛选了13种膦配体,最初的托尔曼锥角 (θ) 特征难以解释反应性的变化。通过特征工程,我们添加第二空间特征、百分比埋藏体积 (%Vbur) 以及电子特征、最小静电势,提升了模型效果(如图1所示)。

图 1. Ni催化的铃木偶联反应的配体研究。(A)交叉偶联反应;(b)多元线性回归反应模型;(c) 托尔曼锥角和埋藏体积的定义。图片来源于Acc.Chem.Res
3. 高维数据集建模
我们试图用机器学习对多种试剂反应数据集建模。数据集包括Buchwald-Hartwig (BH) 胺化数据集,其中含有四个组分的共3955个反应;以及脱氧氟化数据集,含有三个组分的共740个反应(如图2所示)。对于 BH 胺化,模型目标是评估异恶唑对不同芳基和杂芳基卤化物偶联的影响。我们尝试建立预测模型,并希望从特征和机理上解释化学反应。BH 胺化反应用120个DFT的特征描述,而脱氧氟化反应具有23个特征。此外,还对数据集进行了类似的建模研究以预测对映选择性、区域选择性和反应性。

图 2. (A)Buchwald-Hartwig胺化数据集;(b) 醇的脱氧氟化数据集。每个数据集的代表示例。图片来源于Acc.Chem.Res
4. 模型选择
随机拆分数据对模型的泛化能力估计偏乐观,如果目标是预测未知催化剂的产率或为新底物选择产率最高的一组反应条件,则使用启发性的泛化测试更有价值。因此,我们通过留一法交叉验证来估计泛化误差,如下所示:
我们将 BH 胺化反应的试剂以及脱氧氟化反应的醇指定为反应组分,并留出单个分子。数据集包含 22 种BH 胺化反应的异恶唑试剂和 37 种醇,分别产生了 22和 37 个验证集。
我们尝试了线性回归、广义线性模型、支持向量回归、kNN、随机森林、XGBoost 和前馈神经网络,还构建了两个参照模型:使用除标记分子外不携带任何物理信息的随机特征训练一个随机森林;另一个是非随机森林模型,对具有相同底物和条件的所有训练集反应的产率取平均预测产率。我们分析了留一法交叉验证结果(如图3所示)。

图 3. (A)Buchwald-Hartwig胺化数据集的留一法交叉验证结果;(b) 醇的脱氧氟化数据集的留一法交叉验证结果。每个数据集的代表示例。图片来源于Acc.Chem.Res
模型在 BH 胺化数据上的表现显著不同,但是在脱氧氟化数据上的表现相似。这表明在脱氧氟化反应中,泛化误差与模型无关。对于 BH 胺化反应,随机森林的表现明显优于两个参照模型。对于随机森林模型,BH 胺化数据集试剂的均方根误差 (RMSE) 介于 5% 和 25% 之间,而来自脱氧氟化数据集的醇的均方根误差 (RMSE) 则在 9% 和 41% 之间。
原因可能是可能存在模型运行良好的化学空间区域。在图 4 中,每个分子的预测RMSE用PCA降维后使用最主要的两个成分来可视化。对于 BH 胺化试剂,我们发现特征空间的中心区域的预测平均误差低于8% ,而对于脱氧氟化数据,无法确定这样的区域。说明要么基于DFT 的特征化无法捕获脱氧氟化中结构与活性相关的信息,要么该算法无法利用数据集做到这一点。

图 4. (A)BH胺化数据集特征PCA降维后最重要的两个主成分与RMSE的可视化。(B)醇的脱氧氟化数据集特征PCA降维后最重要的两个主成分与RMSE的可视化。图片来源于Acc.Chem.Res
5. 模型训练
如图 5 所示,留一法的交叉验证模型存在过拟合的情况。模型在训练集和验证集之间的差距阻碍了模型对机理的解释。随机森林的有放回采样几乎把所有的样本暴露在了各个树模型中。对于脱氧氟化数据集,每个醇都在有放回抽样中多次抽样,共享相同样本的反应产率;因此,树预测的结果也是相关的,这增强了过拟合。
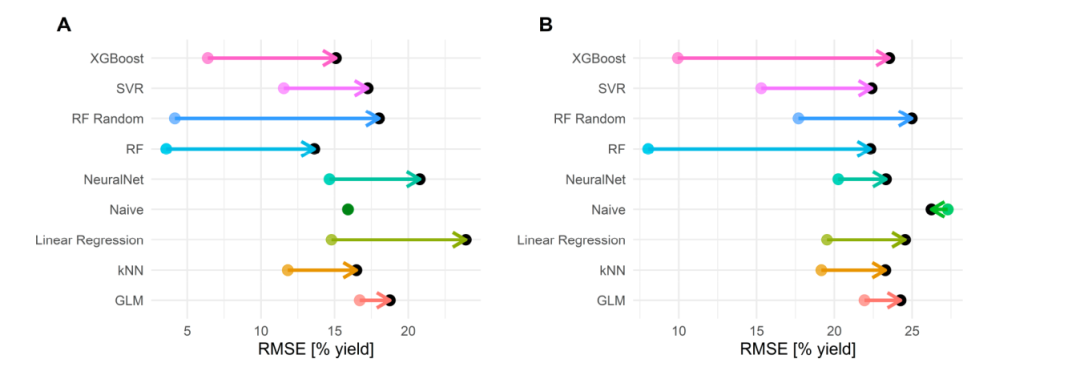
图 5.留一法训练集与验证集上的预测误差对比。彩色点代表训练集误差,黑点代表验证集误差。(A)BH胺化数据集的结果(B)醇的脱氧氟化数据集的结果。图片来源于Acc.Chem.Res
6.模型解释
对于 BH 胺化数据集,使用基于 DFT 的特征和 RF 模型对样本外反应的预测比具有随机特征的模型可信度提高 2%-6%。为了研究分子特征的重要性,我们将特征替换为随机数字,以确保它们无效并在部分噪声化的特征集上训练模型。我们对单个反应成分的所有特征加入噪声,同时保持所有其他试剂的特征不变,由此产生了一些RMSE 增加,但多种试剂的 DFT 特征组合没有增强模型的效果。我们进一步对每个特征进行噪声处理并验证效果,结果发现单个特征的贡献很小,而且大多无关紧要,对单个特征进行干扰并不会降低其整体重要性。最重要的是那些描述 C3 原子的特征,我们在所有数据上训练,逐渐加入C3 NMR位移特征以可视化模型的相关性。在图6中,观察到 C3 NMR位移>150 ppm的试剂产率呈阶梯状增加。从化学结构来看, C3 NMR 位移<150 ppm 的试剂主要具有 C3-H 键;具有 C3 NMR 的试剂位移 >150 ppm,具有完全取代的 C3。带有 C3-H 的异恶唑可以在 N-O 氧化加成后经历 Pd 催化的 Kemp 型重排以形成α-氰基酮和醛(图 6b)。之前的研究表明在没有钯的情况下,即使在加热时也没有观察到异恶唑重排。通过质谱和 NMR 分析,我们发现了钯和苯并 [d]-异恶唑之间的氧化加合物,这表明该加合物可能是图6中异构化的过渡态。虽然不清楚是氧化加合物还是重排产物导致 BH胺化反应中毒,但我们发现由 Pd 催化的试剂重排产生的 3-酮丁酸甲酯衍生物对反应性有较强抑制(图 6c,1-4)。
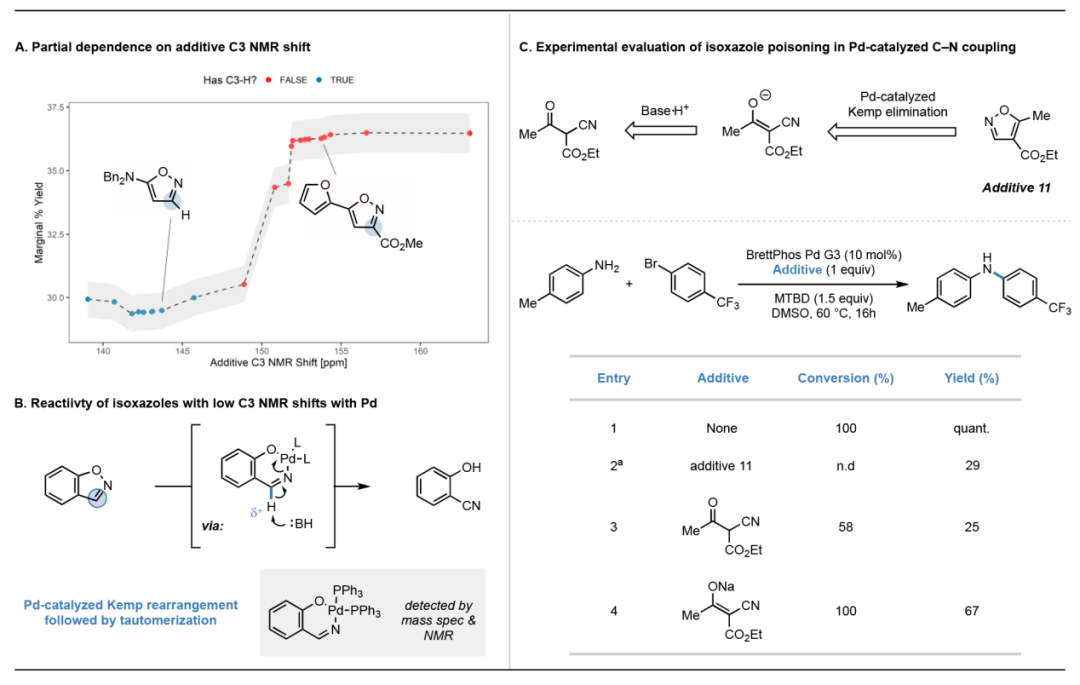
图 6.(A)反应产率与试剂的C3 NMR位移的关系。(B)Kemp-type的重排反应。(C)t-Bu-BrettPhos做配体时的催化剂中毒现象,降低了反应产率。图片来源于Acc.Chem.Res
结论总结
本文介绍的模型可以使用物理不可知特征实现与定量物理特征非常相似的泛化精度。在 BH 数据集中,加入DFT 特征能显著改进模型泛化能力,表明模型具有可迁移的化学能力,并使我们能够了解潜在的机制。
参考文献
Andrzej M. Żuranski, Jesus I. Martinez Alvarado, Benjamin J. Shields, and Abigail G. Doyle,Predicting Reaction Yields via Supervised Learning,Accounts of Chemical Research, 2021, 54, 8, 1856-1865.