

引言
传统的计算方法已在化学反应领域得到广泛应用,例如,QM模拟化学键的生成和断裂,QM/MM和FMO处理涉及生物分子的反应体系等。由于可以计算广泛的数据集,机器学习(ML)已成为对传统计算方法的有力补充。本文侧重于用ML技术解决详尽采样、实验观测值和计算值之间的相关性及如何利用这一点来提高对分子间相互作用的理解和处理整个反应网络的问题。首先介绍了生成模型的结果和实验观察的结果,接着是有机反应中的ML技术,最后概述了ML在整个反应网络中的应用以及前景。
反应可观察值的机器学习
机器学习可用于开发与实验直接相关的可观察指标的综合模型,包括预测反应热、量化反应的速率、态-态截面、起始态到终态的映射,甚至化学反应的产率。
学习反应速率
从微观角度看,在分子尺度上描述反应物和生成物之间转化过程的方法(例如准经典轨线或量子模拟)需要对整个势能面敏感且需要全局反应性的PES。即使对简单的A + BC → AB + C 原子交换反应,通过计算来确定准确的反应速率也是一项艰巨的任务,因为其PES表示非常具有挑战性。目前对气相反应的处理基于a.量化理论下计算数千种能量,b.将这些能量表示为参数化的函数或使用ML技术(如神经网络)再生核希尔伯特空间,c.遵循核动力学使用经典力学QCT或求解三维薛定谔方程。这种方法精度取决于基础PES的准确性。
为了更好地解决这个问题,最近的一项工作使用高斯过程回归来训练校正χ(T)以预测热速率k(T)的可能性:

文章基于共线碰撞做简化处理确定了大约50个不同反应的k(T) ,对13次反应进行了训练,并对模型进行了40次反应测试。用了3~5个描述符来描述χ(T)。与TST或ECK处理相比,机器学习在具有对称和非对称壁垒的系统上表现最佳,学习模型的误差在10%~120%之间,而ECK和TST则分别为80%~180%和180%~760%。
该模型最近被应用于O(3P) + HCl → OH + Cl反应,由于反应势垒大和低能反应共振的存在,这是一个特别具有挑战性的例子,证明了在数据集来源足够广泛质量足够高的前提下,这种方法未来被大量应用是可能的。
态-态模型和速率
从全局看,为N(4S) + NO(2Π) → O(3P) + N2(X1Σg+)反应开发了一个基于NN模型的态-态方法来映射起始态到终态。总的来说,N + NO 和 O + N2通道分别有 6329 和 8733个振动态。对于给定的平移能量Et,通常需要运行104到 105 个QCT 模拟才能收敛一个特定的态-态截面σv , j → v ′ j ′(Et),为所有进入~104终态的~104始态执行此操作需要1012到1013个QCT模拟,这显然是不现实的。为了训练这样的网络,选择了例如内能、振动和旋转量子数或反应物的相对速度这样的特征,为了独立测试最终的NN效果,针对训练中未使用的初始条件,在固定Et下执行了额外的QCT计算。通过实际QCT横截面与预测横截面的比较表明该方法是可行的。为了进一步验证模型,从QCT模拟中明确了初始态选择率并与NN结果进行比较。大多数情况下,相对误差小于5%。一般来说,NN给出的特定状态和温度下的总反应速率与QCT模拟定量上一致。

图1.QCT计算的和NN预测的初始状态选择率之间的相关性。图片来源:Chemical Reviews
反应速率和途径
PES的全局性在高能过程中尤其重要,而与大气或天体物理学相关的过程中的产物数量会极大增加,这使得生成这样的全局PES极具挑战性。为了说明遵循大气相关分子的化学反应问题,作者研究了乙醛AA和烯醇VA的异构化。为此构建了基于NN的反应性PES模型,模拟中激发能量为93.6kcal/mol时,MAE和RMSE分别为0.0071和0.0145kcal/mol。为了验证NN-PES确实允许异构化,使用高达127.6kcal/mol的激发能,此时全局PES的MAE和RMSE分别为0.0132和0.0307kcal/mol。
最近报道了另一种基于ML构建反应式PES并将其用于动力学模拟的方法,它基于置换不变多项式PIP。传统PIP中的展开系数由NN训练。PIP-NN已应用于与大气和燃烧有关的反应,例如HO + CO → H + CO2,主要用于QCT和量子动力学模拟以确定总反应概率、热速率、产物状态振动和旋转分布等。PIP-NN还被用于涉及金属表面的反应性散射,研究体系包括H2/Ag(111)、H2/Co(0001)、H2O/Ni(111)和CO2/Ni(100)。模拟分析表明,PES的垒高度和形状都会影响反应速率,平行和垂直于表面的平移也会对反应速率有影响。最近基于MS-ARMD和NN的全维能量表面的模拟,涉及反应物、中间体和OH消除产物,表明这项技术也可以循序逐步反应。经验FF的拟合精度可以达到约1kcal/mol,而使用PhysNet训练相同的数据的精度可以达到0.02kcal/mol。
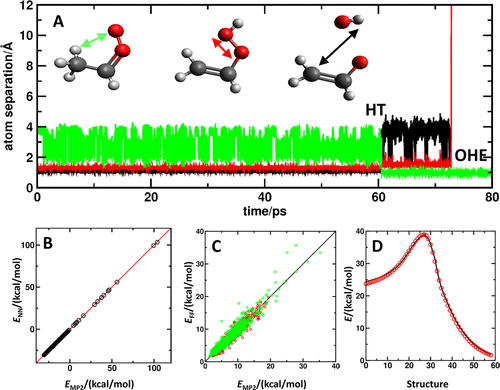
基于ML的方法提供的一种特别有吸引力的可能性是在电子结构理论上的层次上构想模型,并用来自更高层次理论的相当少的数据重新训练它们。这基于一个事实:如果进行了足够准确的计算,给定系统的PES的全局形状在很大程度上保持不变。这些方法通常被称为迁移学习,可以避免高计算成本而且其学习曲线比直接在高级数据上训练独立模型收敛得更快。
ML在反应性生物体系中的应用
反应性生物体系中,蛋白质太大,键断裂和生成的时间尺度太长无法进行完整的ab initio MD模拟。最近,基于ML的能量函数如再生核(RKHS)已被用于跟踪生物体系中的键断裂和生成,一个例子是NO和肌红蛋白的结合。将三维RKHS PES拟合到密度泛函理论计算,用于NO配体相对于血红素单元的径向和角自由度以及铁相对于血红素平面的平面外运动。溶剂化蛋白质-配体系统的所有剩余自由度都用经验能量函数处理。具有这种混合 ML/经验能量函数的广泛反应 MD 模拟提供了 Mb-NO 中亚稳态的第一个结构解释。
实验背景下的机器学习
“逆合成”始于1967年左右。最初基于规则的专家系统预测效果不够好,后来尝试了对标记反应进行训练的机器学习方法,ReactionPredictor使用物化拓扑特征来训练网络,准确率约为96%。基于指纹的神经网络则为80%。使用深度学习优化器可以在40步内找到最佳条件,优化微滴反应所需的总时间为30分钟。最近联合蒙特卡罗树搜索(MCTS)和三个NN(一个用于提出有限数量的自动提取变换,第二个用于预测反应可行性,第三个用于估计每个变换的位置值)的3N-MCTS取得了不菲的成绩。来自2个世界顶级化学机构的45位有机化学家参与了双盲测试,被要求从文献中的实际合成路线和3N-MTCS生成的路线间根据自己的偏好和合成合理性进行选择。在9轮测试中,有6轮化学家们选择了算法建议的路线。尽管取得了这些成就,但使用ML来预测各种有机反应的结果仍然存在局限性,包括天然产物的合成路线、立体化学结果的预测、互变异构的平衡或反应条件的预测。最近,由Chemputer、Chempiler和脚本语言(ChASM)组成的模块化有机合成机器人系统可以驱动四个模块,包括反应瓶、过滤站、液液分离模块和溶剂蒸发模块。该系统用于自动合成化合物,如盐酸苯海拉明、卢非酰胺或西地拉非。除了使标准化学程序自动化并有发现新合成路线的诱人前景外,此类程序还提高了合成程序的可重复性,可将科学家从日常任务中解放出来,从而更专注于产生新想法和有创造性的步骤。
使用质谱数据确定化学结构是专家系统(AI)在涉及分解反应问题的早期应用之一。最近,开发了基于神经网络的技术来解决电子电离竞争性碎裂建模(CFM-EI)的问题。对于给定的化学结构,该模型可以预测电子电离(EI)质谱(MS)。训练NN所需的化学特征包括断键类型、相邻键类型、官能团特征等属性。用于训练、测试和验证的数据集包含约2000个分子。当查询测量的参考光谱时,该模型的性能为 77%,查询 NIST 数据库时为 43%。化合物结构鉴定(CSI)在预测指纹和鉴定代谢物(FingerID)中使用分子片段树和基于多核学习的分子指纹预测。对约 6200 种化合物进行训练的结果显示正确识别率约为 30%。在基于 PubChem 的比较分析中,CSI:FingerID 的识别率约为32%,而 CFM-ID 的识别率约为12%。
整个反应网络的机器学习
反应网络与化学的各个分支相关,包括但不限于大气反应、燃烧以及天体物理和生物体系。通常通过求解大量耦合常微分方程,在随机网络级别对此类网络进行采样。最近,提出了一种基于 NN 的模型来跟踪空间和时间上的燃烧反应。这些模拟使用 DeepMD NN 架构来计算 3000 K 下甲烷燃烧(起始 100 个CH4和 200 个O2分子)的能量和力,并发现了 798 种不同的化学反应,其中一些目前未知。涵盖的总模拟时间以纳秒为单位,这些模拟的准确性仅受神经网络训练的电子结构数据的限制。在最近的另一项研究中,使用 ML 训练的原子化能量模型模拟甲烷燃烧,用核岭回归(KRR)和原子位置平滑重叠(SOAP)来表示,在该模拟中形成了几个值得注意的物质,包括甲醇、甲酸和 Criegee 中间体。
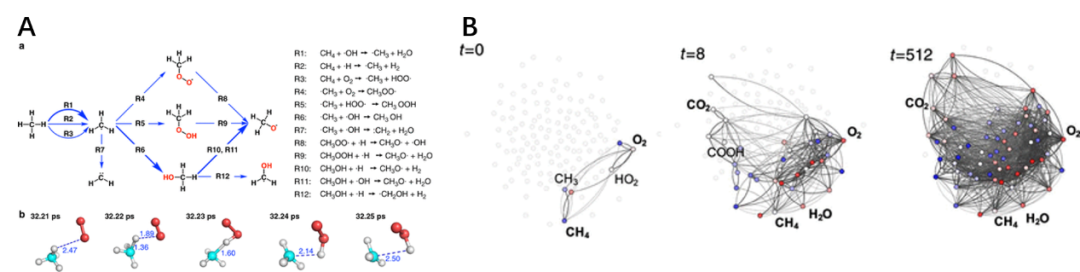
未来发展
对小体系(包含很少的原子)的一个挑战是,当采用ML技术时,是否可以显著减少用于构建全局反应性PES的参考点数量,以便准确表示分子间PES。所需的点数可能取决于(置换)对称性的存在和所涉及的化学物质。未来贝叶斯优化技术的一个有趣应用是将其与Δ-ML、迁移学习或基于实验确定的光谱或反应散射数据的变形相结合。除了点数之外,还需要考虑系统的哪些配置用于参比计算的问题,这些点应该放在信息量最大的区域。另一个角度是学习高质量的PES,这又涉及对从实验观察到的高质量PES的探索和改进。该领域还有一个挑战是缺乏统一准确和有效的方法来求解电子薛定谔方程。
未来发展的另一个领域涉及电子激发态的反应性,对较大的体系,激发态的动力学是一个极有挑战性的问题,应用于此的ML技术刚刚才开始出现,需要特别关注的是非绝热动力学和相邻电子状态之间转换中涉及的耦合矩阵元素。
预计将目前可用的 ML 技术用于使用质谱法进行片段化与从头计算和来自现有数据库的数据相结合将进一步促进 MS 实验中化学结构测定的定量方面。
基于机器学习特别是深度学习的方法无法得到更广泛应用的主要障碍之一,是机器学习的问题处理结果与化学推理之间的薄弱环节,即“因果关系”问题。药物设计中的另一个挑战是药物活性化合物的数据库质量和结构多样性。与从序列预测蛋白质结构的问题相反,药物活性物质的设计和开发需要了解“为什么”配体上的特定修饰对其生理作用有益。同样,对于实际“蛋白质折叠途径”和速率的问题,预计需要对实际过程有更深入的了解,这与 alphafold 解决的“终点问题”不同。
总之,应用于化学反应的基于机器学习的方法是一个快速扩展的领域。未来的挑战涉及准确、定量和详尽地确定反应结果、速率和(内部)状态分布。结合机器人平台,可以使用 ML 和贝叶斯技术优化反应产率和反应条件。在酶设计领域,结合实验与基于 ML 的方法对于蛋白质-配体相互作用和识别,将对蛋白质结构预测的最新进展提供重要的见解。
参考文献:
Meuwly M. Machine Learning for Chemical Reactions. Chem Rev 2021,doi: https://doi.org/10.1021/acs.chemrev.1c00033