

引言
在最近的CASP14上,DeepMind给出了非常精确的预测(即AlphaFold 2)。本文作者探索了结合相关思想的网络体系结构,并在一个三轨网络中获得了最佳性能,其中一维序列级、二维距离图级和三维坐标级的信息进行依次转换和集成。三轨网络产生的结构预测精度接近CASP14中的DeepMind,能够快速解决具有较高难度的X射线晶体学和冷冻电镜结构建模问题。该网络还能够仅从一维序列信息快速生成精确的蛋白质-蛋白质复合物模型,优化传统建模方法(即需要对单个亚单位进行建模,然后进行对接)。作者开源此方法以加速生物学研究。有了RoseTTAFold,在一台游戏电脑上只需10分钟就可以计算出蛋白质结构。这项工作由贝克实验室博士后学者Minkyung Baek领导,相关的研究成果以“Accurate prediction of protein structures and interactions using a three-track neural network”为题发布在国际著名杂志Science上。
网络结构开发
受一些研究成果的启发,以提高结构生物学研究中蛋白质结构的准确预测和推进蛋白质设计为目标,作者探索了不同组合的网络结构。最终成功地制造了一个“双轨”网络,信息沿着一维序列比对轨道和二维距离矩阵轨道并行流动,其性能远远优于trRosetta(在CASP14中仅次于AlphaFold2)。作者通过扩展到在三维坐标空间中运行的第三个轨道,在序列、剩余距离和方向以及原子坐标之间提供更紧密的连接,实现更好的性能。RoseTTAFold是一个“三轨”神经网络,意味着它同时考虑蛋白质序列中的模式、蛋白质的氨基酸如何相互作用以及蛋白质可能的三维结构。在这种结构中,一维、二维和三维信息来回流动,使网络能够共同推理蛋白质的化学部分与其折叠结构之间的关系。
三轨模型在CASP14目标上的性能仍然不如AlphaFold2,这可能反映了作者可以使用的硬件限制了探索的模型性能。在对少于400个残基的蛋白质进行结构预测时,在序列和模板搜索(约1.5小时)之后,端到端版本的RoseTTAFold在RTX2080 GPU上约10分钟可以完成预测,pyRosetta版本需要5分钟进行网络计算,需要1小时在15个CPU核上进行所有原子结构生成。由于计算机内存限制和忽视侧链信息而导致的不完全优化可能解释了端到端版本与pyRosetta版本相比性能较差的原因。作者期望,一旦计算机硬件的局限性被克服,端到端的方法最终会同样准确。
辅助实验蛋白结构测定
作者同样也研究了RoseTTAFold的实用性,便于辅助X射线晶体学和低温电子显微镜进行实验结构测定,为目前未知结构的关键蛋白质提供帮助。通过分子置换(MR)解决X射线结构通常需要相当准确的模型。RoseTTAFold的精确度比现有方法高得多,这使得作者可以使用它来解决具有挑战性的MR问题,并改进解决方案。四个晶体数据集,使用RoseTTAFold模型重新分析:一种来自牛的甘氨酸N-酰基转移酶(GLYAT),一种细菌氧化还原酶,一种细菌表面层蛋白(SLP)和来自真菌黄孢原毛平革菌的分泌蛋白Lrbp。在所有情况下,预测模型都与真实结构具有足够的结构相似性,从而给出成功的MR解决方案。此外,在使用RoseTTAfold预测GBD结构的过程中,尽管局部分辨率很低,但预测的结构仍然很容易拟合到电子密度图中。
蛋白-蛋白复合物结构预测
为了探索RoseTTAFold对三条链以上的复合物的预测,作者使用它来生成人类IL-12R/IL-12复合物模型(四条链)。先前公布的IL-12受体复合物的冷冻电镜图谱显示与IL-23受体相似的拓扑结构;然而,分辨率不足以观察IL-12Rβ2和IL-12p35(34)之间的详细相互作用。这种认识对于剖析IL-12和IL-23的具体作用以及发现可以产生阻断IL-12而不影响IL-23信号传导的抑制剂的研发是有重要意义的。RoseTTAFold模型很好地拟合了实验cryo-EM 密度图,并确定了IL-12p35中的Y189和IL-12Rβ2中的G115之间的相互作用,类似于IL-23p19中的W156和IL-23R中的G116之间的堆积。此外,该模型还观察到一些在IL-12冷冻电镜结构中未观察到的相互作用(IL-12Rβ2 D26可能与IL-12p35中附近的K190和K194相互作用)。
图表汇总

图1. RoseTTAFold的网络架构和性能。图片来源于Science
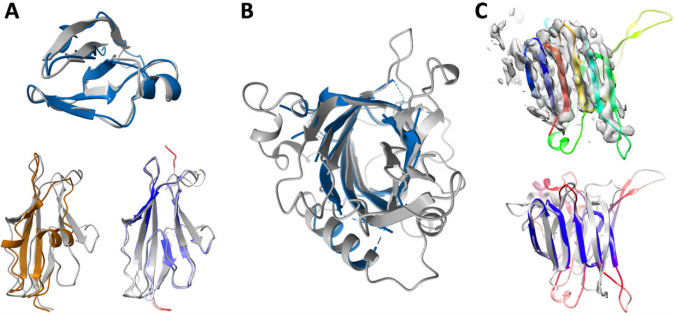
图2. RoseTTAFold预测的蛋白结构。图片来源于Science
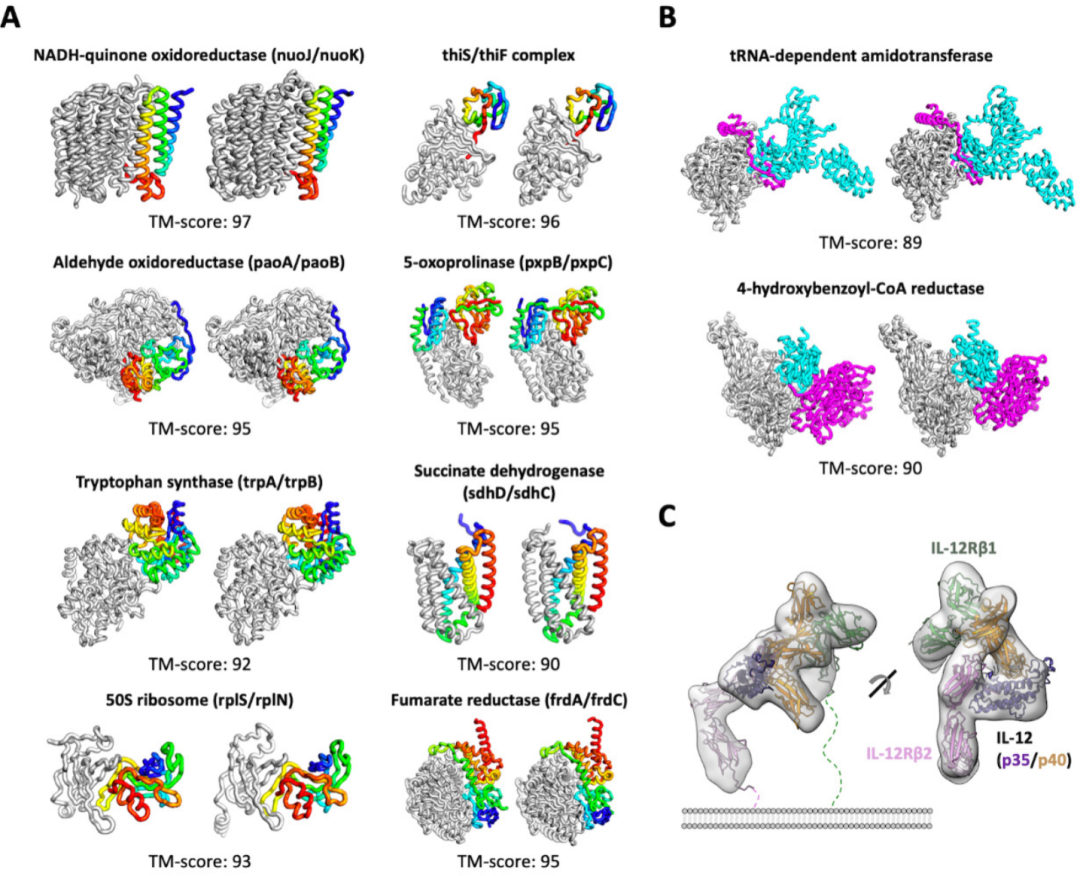
图3. RoseTTAFold预测的蛋白-蛋白复合物结构。图片来源于Science
结论总结
RoseTTAFold能够解决具有挑战性的X射线晶体学和冷冻电镜建模问题,在没有实验结构的情况下提供对蛋白质功能的洞察,并快速生成蛋白质-蛋白质复合物的精确模型。对蛋白质-蛋白质复合物数据集的进一步训练将可能进一步变为多蛋白组装的建模。该方法可以与现有的小分子抑制剂和新型蛋白设计方法相结合,以改进现有计算手段。三轨结构在对序列、距离和坐标信息处理的同时为低温电镜结构处理以及蛋白质设计等问题打开了新的大门。
Public Server和代码下载
RoseTTAFold Public Server:
https://robetta.bakerlab.org
RoseTTAFold GitHub: https://github.com/RosettaCommons/RoseTTAFold
参考文献
Minkyung Baek, Frank Dimaio, Ivan Anishchenko, et al., Accurate prediction of protein structures and interactions using a three-track neural network, Science, 2021. DOI: 10.1126/science.abj8754.