
引言
通过整合深度学习和共进化分析,从蛋白质的一级序列来预测其三级结构方法已经有了很大的改进,在CASP13和CASP14中表现的极为明显。本文描述了一项最新研究,分析了网络大小和共进化数据的有效性及其在天然蛋白和人工设计蛋白上的表现。结果表明,一个大的ResNet(卷积残差神经网络)可以预测出26个正确的折叠结构,精度超过80%。当不使用共进化时,ResNet仍然可以预测18个正确结构,大大超过了以前不使用共进化的方法。即使只有一级序列,ResNet也能预测出所有人工设计蛋白质的正确折叠结构。此外,ResNet在没有共进化信息的情况下要比有共进化信息的情况下表现的更好。这些结果表明ResNet可以学习蛋白质序列-结构之间的重要关系。这对蛋白质设计和工程有重要的意义,尤其是在共进化数据不可用时。
方法概述
本文方法由两个关键部分组成。第一个部分是用于预测三个主链原子对(C–C、C–C和N–O)的离散概率分布以及残基间取向(trRosetta定义)的深度ResNet。第二个部分是一个增强的基于梯度的能量最小化方法,该方法从预测的距离、方向和主干扭转角得到的电势来建立蛋白质模型。PyRosetta fast relax方法用于侧链packing和减少空间冲突。整体网络架构与CASP13中使用的网络架构类似,除了这里使用的ResNet更大更宽,具有100个2D卷积层,并且平均每层150个滤波器。CASP13中的ResNet只有60个2D卷积层,平均每层80个滤波器。本文使用多任务学习策略,并进行了充分的测试,可以同时预测所有的距离和方向矩阵。多任务学习并不能获得明显的性能提升,但它大大减少了模型训练的时间和模型的数量。
采用共进化信息时建模精度
通过为每个目标生成150个诱饵并对它们进行聚类来测试本文ResNet。当四个ResNet模型作为一个集合时,第一和最佳预测模型的平均质量值(TMscore)分别为0.638和0.659。这一点可以进一步改进。每个目标生成600个诱饵可将第一和最佳模型质量值分别提高到0.640和0.675,并且将2D ResNet大小增加到120个卷积层(和每层170个滤波器)可将第一和最佳模型质量值分别提高到0.646和0.673。在考虑最佳模型的情况下,本文方法预测了32个FM目标中26个的正确折叠结构。这些结果表明,ResNet可以生成新颖的结构,产生的模型质量明显高于简单训练集所能达到的效果。如下图所示,模型质量与MSA深度之间存在适度相关性。本文方法可以预测出所有测试目标的正确折叠,但两个测试目标[ln(MSA深度)>3]除外。本文的三维建模精度要比之前报道的结果效果更好。例如,在CASP13中,AlphaFold 1预测了32个CASP13 FM目标中23个的正确折叠结构,第一和最佳模型的平均TMscore 分别为0.583和0.625。trRosetta对32个CASP13 FM目标建模的平均TMscore 为0.618。
不使用共进化信息时预测三维模型的精度
在这里,本文总结和分析了没有使用共同进化的ResNet如何预测32个CASP13 FM靶点和21个人类设计的蛋白质的结构。没有共进化信息的CASP13 FM靶模型。ResNet生成的第一和最佳三维模型的平均质量(TMscore)分别为0.478和0.506。当考虑到最佳预测模型时,ResNet预测了32个CASP13 FM目标中的18个的正确折叠。图2c显示了建模精度和目标-训练结构相似性之间的弱相关性(相关系数=0.363,趋势线R=0.1315)。建模精度与MSA深度(相关系数=0.211,趋势线R=0.0448)之间的相关性比使用共进化时弱,这并不意外。为了让读者更好地理解本文的工作,作者将其与顶级服务器Robetta和RGN进行了比较。Robetta是CASP13中最好的服务器,它没有使用深度学习。它结合了基于模板的建模、从头计算和基于共进化的接触预测。RGN是一种端到端的深度学习方法,它可以从序列中预测蛋白质结构,而不是共进化。Robetta提交的32个CASP13 FM目标的first和best模型的平均核心值分别为0.390和0.430。RGN模型的平均核数为0.251。Robetta和RGN都比本文的方法差很多。此外,对于几乎所有32个FM目标,ResNet在没有共进化信息的情况下训练预测出比RGN质量更好的3D模型。RGN无法预测所有32个测试目标的正确折叠结构,而Robetta和ResNet则只是分别预测了7个和15个正确折叠,即使只考虑第一模型。模拟人类设计的蛋白质。ResNet可以预测18种设计蛋白质中16种的正确折叠结构。当进化信息完全不可用时,ResNet仍然可以预测几乎所有21种蛋白质的正确折叠结构。由RGN生成的21个模型的平均TMscore为0.363,比没有共同进化和使用原始序列作为输入的ResNet训练的要差得多。总的来说,RGN只预测了三种人类设计的蛋白质的正确折叠,而ResNet只预测了14种人类设计的蛋白质的正确折叠。Yang等人测试了21种人类设计的蛋白质中的11种。trRosetta预测的3D模型的平均TMscore为0.661,与使用共进化训练的ResNet相当,这并不奇怪,因为trRosetta也使用共进化训练。
图表汇总

表1. 不同配置下ResNet对CASP13目标进行长距离接触预测的精度和F1值
表格来源于Nat. Mach. Intell.

图1. 不同ResNet模型对31个CASP13 FM 目标的接触预测精度
图片来源于Nat. Mach. Intell.

图2. 32个CASP13 FM 目标的3D建模精度(Tmscore)
图片来源于Nat. Mach. Intell.
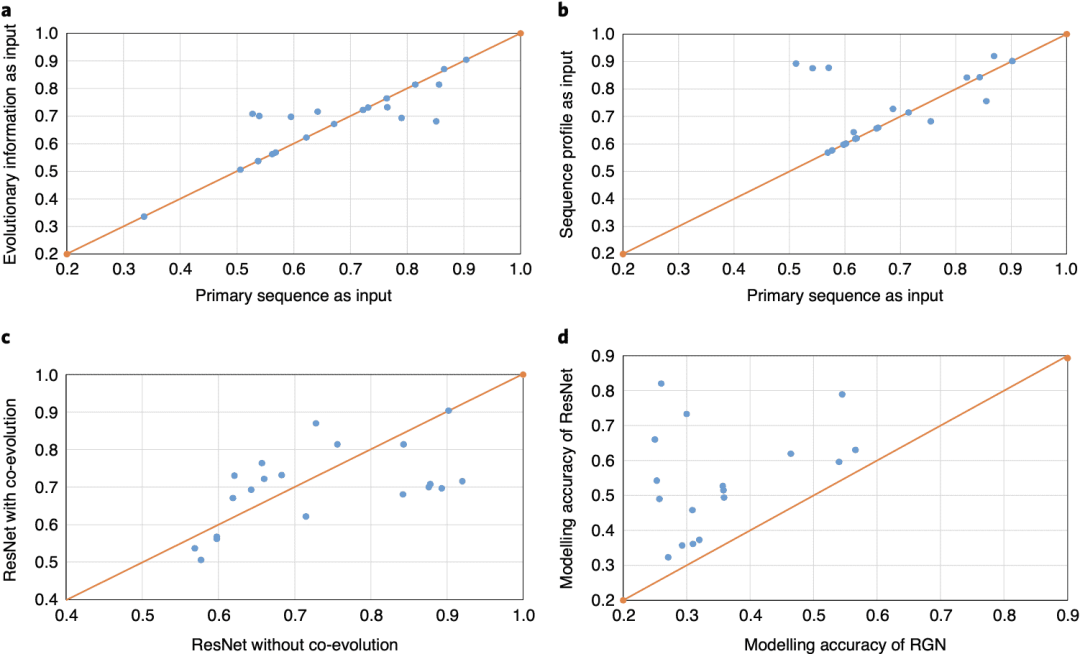
图3. 对于人工设计蛋白质的三维建模精度
图片来源于Nat. Mach. Intell.
亮点总结
本文介绍了deep-ResNet在结构预测方面的最新研究成果。研究表明,虽然ResNet在结构预测方面有了很大的改进,但是仍然可以改进。可以使用一个更大的ResNet、共进化矩阵和元基因组数据的组合来进行优化。此外,基于梯度的能量最小化和内部残基方向也有助于三维建模。
当不使用共进化时,用序列训练的ResNet可以预测超过一半的CASP13 FM靶点和测试的所有人类设计的蛋白质的正确折叠结构。此外,对于人类设计的蛋白质,当序列同源物较少时,序列轮廓可能比共进化更有用。在进化信息不可用的情况下(即只有一级序列可用),ResNet仍然可以预测几乎所有人类设计的蛋白质的正确折叠,尽管平均模型质量有所下降。这些结果表明ResNet可以从实验蛋白结构中学习控制蛋白质折叠的重要信息。因此,ResNet可用于快速验证设计的蛋白质是否具有所需的结构,从而避免了对昂贵且耗时的湿实验室的需要。
尽管ResNet可以在不使用任何进化信息的情况下预测人类设计的蛋白质的正确折叠结构,但它在天然蛋白质上的效果并不好,可能是因为人类设计的蛋白质通常比天然蛋白质有更深的能量井。对于大多数天然蛋白质,本文方法仍然需要少量的序列同源物才能工作。然而,在自然界中,蛋白质在不知道其序列同源物的情况下折叠。希望在将来可以得到一种方法能够在不使用序列同源物的情况下折叠蛋白质。
代码下载
https://github.com/j3xugit/RaptorX-3DModeling/
https://doi.org/10.5281/zenodo.4642250
在线服务
http://raptorx.uchicago.edu/
参考文献
Xu, Jinbo; McPartlon, Matthew; Li, Jin, Improved Protein Structure Prediction by Deep Learning Irrespective of Co-Evolution Information, Nature Machine Intelligence, 2021, ASAP. DOI: 10.1038/s42256-021-00348-5.